Général
L’augmentation de l’activité de TREX1 booste la recombinaison homologue médiée par CRISPR-Cas9 !
Main Repair of CRISPR–Cas9-induced double-strand breaks (DSBs) occurs by one of two main mechanisms: non-homologous end joining (NHEJ) or homology-directed repair (HDR)1,2,3. The HDR pathway is used to introduce sequences of up to 2–3 kilobases (kb) into the genome that are supplied on an exogenous DNA template. In most human cells, the efficiency of HDR
Réparation des Cassures Double Brin Induites par CRISPR-Cas9
La réparation des cassures double brin (DSB) induites par CRISPR-Cas9 se fait principalement par deux mécanismes : la jonction non homologue des extrémités (NHEJ) et la réparation dirigée par homologie (HDR). La voie HDR permet d’introduire des séquences allant jusqu’à 2 à 3 kilobases (kb) dans le génome, fournies par un modèle d’ADN exogène. Cependant, dans la plupart des cellules humaines, l’efficacité de la HDR est très inférieure à celle de la NHEJ. Le contexte cellulaire joue un rôle crucial dans l’efficacité de la HDR, qui est limitée au cycle cellulaire et est particulièrement active durant les phases S/G2. L’expression différentielle, le contexte cellulaire et/ou la charge mutationnelle peuvent également influencer cette efficacité. Le ciblage du même locus par CRISPR-Cas9 avec des réactifs identiques dans différents types cellulaires peut entraîner des variations d’efficacité, allant de 30 % des allèles à une inactivité totale. L’efficacité de l’édition par prime editing dépend de l’état de réparation des mésappariements des cellules ciblées, mais un biomarqueur similaire pour l’efficacité de la HDR médiée par CRISPR-Cas9 reste à identifier.
Impact des Mutations sur l’Édition Génétique
Les cellules des patients présentant des mutations dans les gènes de réparation de l’ADN peuvent également compromettre l’édition génomique médiée par CRISPR-Cas9, rendant difficile la correction des troubles ciblés. Par exemple, la perte de fonction d’un des 22 gènes impliqués dans l’anémie de Fanconi (FA) peut largement empêcher la HDR. La FA est un trouble génétique rare caractérisé par une défaillance de la moelle osseuse et une prédisposition aux cancers à un âge avancé. Les tentatives de correction des mutations chez les patients atteints de FA par HDR induite par CRISPR-Cas9 ont montré des efficacités très faibles, limitant les approches d’édition génomique potentiellement curatives à celles qui contournent la HDR, mais qui ne sont pas applicables à tous les allèles de la FA.
Identification des Facteurs Limitants de la HDR
Dans cette étude, nous avons réalisé un criblage à l’échelle du génome dans des lignées cellulaires lymphoblastiques (LCL) dérivées de patients atteints de FA pour identifier les facteurs qui restreignent la HDR médiée par CRISPR-Cas9. Nous avons découvert que TREX1, une nucléase associée au réticulum endoplasmique (RE) impliquée dans l’immunité innée, joue un rôle majeur dans la réduction de la HDR induite par CRISPR-Cas9 dans les cellules humaines. L’invalidation de TREX1 restaure la HDR dans les cellules dérivées de patients atteints de FA et dans des modèles cellulaires couramment utilisés avec une efficacité de HDR naturellement faible. La protection chimique des modèles d’ADN donneurs de manière à prévenir l’activité de TREX1 permet de restaurer la HDR dans ces cellules exprimant TREX1 à plusieurs loci. Nos travaux soulignent l’importance des facteurs cellulaires dans la régulation des résultats de l’édition génomique et offrent une explication rationnelle de l’efficacité apparemment stochastique de la HDR médiée par CRISPR-Cas9 dans divers contextes cellulaires, tout en proposant une voie potentielle vers des niveaux élevés d’édition génomique dans de nombreux modèles cellulaires et lignées cellulaires primaires limités par l’expression de TREX1.
Résultats de l’Étude
Réactivation de la HDR par Élimination de TREX1 dans les Cellules de Patients atteints de FA
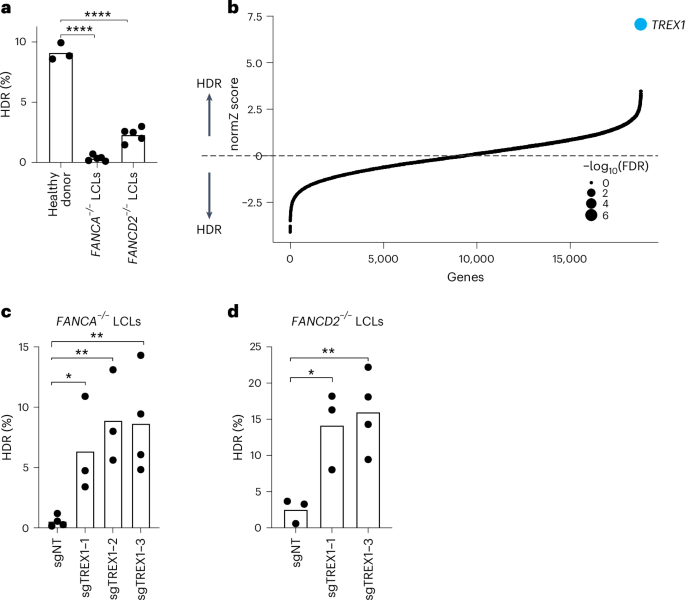
Nous avons d’abord analysé les efficacités de la HDR médiée par CRISPR-Cas9 dans des LCL provenant de patients FANCA et FANCD2 en utilisant un rapporteur BFP-à-GFP précédemment publié. Cette séquence BFP peut être ciblée par un ribonucléoprotéine (RNP) Cas9 et un oligonucléotide simple brin (ssODN) approprié pour convertir BFP-His151 en GFP-Tyr151. Après avoir optimisé les conditions d’électroporation dans des LCL de donneurs sains, nous avons constaté que les efficacités de HDR dans les LCL de FA étaient extrêmement faibles par rapport aux LCL de type sauvage, en particulier dans le contexte FANCA (par exemple, 0,3 ± 0,2 % FANCA contre 2,3 ± 0,6 % FANCD2 contre 9,1 ± 0,7 % de type sauvage). Nous avons donc cherché des facteurs dont l’élimination pourrait restaurer la HDR dans les cellules FANCA.
Exploration des Mécanismes de Réparation de l’ADN par CRISPR-Cas9
Dans notre étude, nous avons utilisé un criblage génétique combiné pour identifier les éléments qui régulent la réparation par homologie de recombinaison (HDR) médiée par CRISPR-Cas9. En nous basant sur un rapporteur HDR précédemment publié, nous avons mis en œuvre un système d’inhibition CRISPR (CRISPRi) pour réduire l’expression d’un gène par cellule tout en induisant une cassure double brin (DSB) et en fournissant un modèle de réparation ssODN au locus du rapporteur. La séparation des cellules par tri cellulaire par fluorescence (FACS) a permis de détecter la conversion de BFP en GFP, et le séquençage des ARN guides récupérés a révélé les facteurs influençant la HDR. Nos travaux antérieurs étaient limités à un ensemble de 2 000 gènes centraux liés au métabolisme de l’ADN, mais cette fois, nous avons élargi notre approche à l’échelle du génome dans un contexte cellulaire dérivé de patients.
Ingénierie des Cellules LCL pour l’Étude de la HDR
Nous avons d’abord modifié des lignées cellulaires lymphoblastoïdes (LCL) FANCA par lentivirus pour exprimer de manière stable KRAB-dCAS9-mCherry (CRISPRi). Tous les ARN guides (gRNAs) de la bibliothèque CRISPRiv2 ont été reclonés dans un vecteur lentiviral modifié, avec BFP en aval du cassette sgRNA. Lors du criblage, l’électroporation de Cas9 RNP et de ssODN dans les LCL CRISPRi FANCA a produit une fraction GFP+ d’environ 0,5 %. Nous avons utilisé une stratégie de double seuil pour isoler rapidement les cellules rares présentant des niveaux élevés de HDR. Les ARN guides ont été amplifiés par PCR à partir des populations non triées et GFP+, puis séquencés par séquençage de nouvelle génération (NGS) pour quantifier les abondances et calculer les scores d’enrichissement au niveau des gènes.
Identification de TREX1 comme Facteur Clé de la HDR
Nous avons découvert que le gène TREX1 était fortement enrichi dans la population HDR GFP+ (taux de fausse découverte (FDR) de -7). TREX1 est une exonucléase 3′-5′ ancrée à la membrane externe du réticulum endoplasmique, jouant un rôle crucial dans la suppression de l’activation chronique de la synthase de GMP-AMP cyclique (cGAS) lors de la réponse immunitaire innée à l’ADN cytosolique. TREX1 agit sur les molécules d’ADN simple et double brin, mais présente une activité bien moindre envers l’ARN et les hybrides ARN-ADN. Des mutations dans TREX1 sont associées à des troubles auto-immuns, tels que le syndrome d’Aicardi-Goutières.
Validation des Résultats de Criblage
Pour confirmer nos résultats initiaux, nous avons cloné plusieurs gRNAs ciblant TREX1 et réalisé des expériences CRISPRi dans les LCL FANCA et FANCD2. L’analyse de l’expression de TREX1 par qRT-PCR a montré une expression très élevée dans les lignées cellulaires FA par rapport aux cellules de donneurs sains. Le knockdown de TREX1 a restauré l’activité HDR dans les contextes cellulaires des patients FANCA et FANCD2.
Impact de TREX1 sur la Réparation de l’ADN
Nous avons également créé un knockout isogénique de TREX1 dans des cellules RPE-1 hTERT, qui possèdent par ailleurs des mécanismes de réparation de l’ADN fonctionnels. L’expression ectopique de TREX1 sauvage dans le clone knockout a révélé que les cellules RPE1 sauvages réalisent des niveaux modérés de HDR induite par CRISPR-Cas9, tandis que le knockout de TREX1 a considérablement augmenté l’activité HDR. De plus, la surexpression de TREX1 sauvage dans le contexte knockout a presque complètement inhibé la HDR.
Interaction de TREX1 avec les Modèles de Réparation
Nous avons observé que TREX1 interagit avec les modèles HDR ssODN et que cette interaction est inhibée par la protection phosphorothioate. Les donneurs HDR ont été introduits par électroporation dans les cellules RPE-1 hTERT, et après 20 minutes, les cellules ont été collectées pour préparer des lysats.
Impact de TREX1 sur l’efficacité de HDR dans les cellules humaines
Dans le cadre de l’immunoprécipitation avec des billes de streptavidine, des analyses ont été réalisées en utilisant des anticorps anti-TREX1, anti-RPA32 et anti-β-actine. L’incorporation de quatre liaisons phosphorothioates (PT) aux extrémités 5′ et 3′ ou uniquement à l’extrémité 3′ d’un ssODN a permis d’améliorer l’efficacité de la réparation par homologie (HDR) dans des lignées cellulaires FANCA-/- et FANCD2-/- ainsi que dans des cellules RPE1. La protection à l’extrémité 5′ se comporte comme un ssODN non protégé. L’efficacité de HDR a été mesurée à l’aide d’un essai BFP-to-GFP, chaque point représentant un réplicat biologique individuel.
Interaction de TREX1 avec les ssODN
Étant donné le rôle connu de TREX1 en tant qu’exonucléase, il est possible qu’il modifie les profils de réparation au niveau de la rupture ou qu’il interagisse avec le modèle de réparation de l’ADN utilisé lors de la HDR médiée par CRISPR-Cas9. Nous avons mené des expériences d’édition dans des cellules HeLa de type sauvage et dépourvues de TREX1, en analysant les variations d’indels autour de sites de DSB potentiels. Cependant, aucune variation significative dans la formation d’indels, en particulier de grandes délétions, n’a été observée.
Nous avons ensuite examiné si TREX1 interagissait physiquement avec un ssODN introduit dans les cellules. Nous avons électroporé des cellules RPE-1 hTERT de type sauvage et TREX1-/- avec un modèle ssODN biotinylé à l’extrémité 5′ et avons effectué une immunoprécipitation après deux heures. En tant que contrôle positif, nous avons détecté RPA32, qui interagit fortement avec les ssODNs. RPA32 était facilement mesurable dans les échantillons immunoprécipités des deux types de cellules. TREX1 était également fortement associé au ssODN biotinylé, indiquant que TREX1 interagit avec les modèles d’ADN électroporés et suggérant qu’il pourrait réduire leur disponibilité pour la HDR.
Localisation et rôle de TREX1 dans la réparation de l’ADN
TREX1 est une enzyme associée au réticulum endoplasmique, avec son domaine nucléase orienté vers le cytosol. Il est donc probable que TREX1 dégrade les modèles d’ADN cytoplasmiques avant leur diffusion dans le noyau. Cependant, certaines études ont suggéré que TREX1 pourrait jouer un rôle actif dans la réparation de l’ADN et se déplacer vers le noyau après des dommages à l’ADN. Nos données, utilisant des ssODNs protégés, indiquent que le rôle principal de TREX1 dans la restriction de la HDR médiée par CRISPR-Cas9 est lié à la disponibilité des modèles. Des études récentes ont également montré que TREX1 s’associe à l’ADN transfecté dans le cytosol. Pour explorer si la localisation de TREX1 était affectée lors de l’édition par CRISPR-Cas9, nous avons analysé sa localisation 24 heures après plusieurs formes d’édition. Nous avons constaté que TREX1 restait en dehors du noyau dans toutes les conditions, ce qui suggère qu’un DSB médié par Cas9 n’est pas suffisant pour induire des changements significatifs dans la localisation de TREX1.
Protection des ssODN et efficacité de HDR
Si TREX1 est effectivement un exonucléase majeur affectant la stabilité des modèles ssODN, un ssODN protégé à l’extrémité 3′ devrait bloquer son activité hydrolytique. Nous avons donc mesuré l’efficacité de HDR en utilisant des ssODNs protégés ayant des liaisons phosphorothioates entre cinq nucléotides aux extrémités 5′ et/ou 3′. Un ssODN non protégé a montré une efficacité modérée de HDR dans les cellules RPE-1 de type sauvage et une très faible efficacité dans les lignées FANCA-/- et FANCD2-/-. La protection du ssODN aux deux extrémités a considérablement augmenté l’efficacité de HDR dans tous les types cellulaires, sans affecter les profils d’indels moléculaires. En accord avec l’activité de TREX1, la protection à l’extrémité 3′ était suffisante pour augmenter l’efficacité de HDR, tandis que la protection à l’extrémité 5′ se comportait de manière similaire à un ssODN non modifié.
Variabilité de l’efficacité de HDR selon les lignées cellulaires
Nous nous sommes interrogés sur la possibilité que TREX1 puisse expliquer la variabilité de l’efficacité de HDR médiée par CRISPR-Cas9 observée dans différentes lignées cellulaires humaines. L’expression de TREX1 varie considérablement entre les lignées cellulaires, ce que nous avons confirmé par qRT-PCR. Les lignées cellulaires couramment utilisées pour l’édition génomique, telles que K562 et HEK293, présentent de faibles niveaux de TREX1, tandis que des cellules où la HDR est plus difficile, comme U2OS et Jurkat, expriment des niveaux élevés de TREX1. Nous avons testé cette observation de plusieurs manières.
Pour déterminer si l’expression de TREX1 est un indicateur prédictif de l’efficacité de HDR médiée par CRISPR-Cas9, nous avons testé un rapporteur intégré BFP-to-GFP dans les cellules Jurkat, MDA-MB-231, U2OS, HeLa et K562. Les cellules HeLa, qui expriment des niveaux élevés de TREX1 selon le Human Protein Atlas, ont montré une faible efficacité de HDR. Notamment, l’efficacité de HDR dans les cellules HeLa peut être restaurée au même niveau que les cellules K562 par un knockdown de TREX1 ou l’utilisation d’un ssODN protégé. Parmi les lignées cellulaires testées, seules les cellules K562 à faible expression de TREX1 ont montré une haute efficacité de HDR avec un ssODN non protégé. Les ssODNs protégés ont augmenté l’efficacité de HDR dans toutes les cellules exprimant TREX1, mais n’ont pas davantage augmenté l’efficacité dans les cellules K562 à faible TREX1.
Impact de la Protection des ssODN sur l’Efficacité de l’HDR
Introduction à l’HDR et aux ssODN
L’édition génétique par CRISPR-Cas9 a révolutionné la biologie moléculaire, permettant des modifications précises de l’ADN. Cependant, l’efficacité de la réparation par homologie dirigée (HDR) peut varier considérablement selon les types cellulaires. Des études récentes ont montré que l’expression de la protéine TREX1 peut inhiber l’HDR dans certaines cellules, mais l’utilisation de ssODN protégés pourrait améliorer cette efficacité.
Résultats des Tests sur l’Efficacité de l’HDR
Nous avons examiné l’impact de la protection des ssODN sur l’introduction de divers types de mutations, y compris des changements de nucléotides uniques, des insertions courtes et des délétions, dans des cellules exprimant TREX1, telles que les cellules RPE1 et U2OS. Les résultats ont été mesurés par séquençage de nouvelle génération (NGS). L’utilisation de ssODN non protégés a révélé une efficacité d’HDR relativement faible dans presque tous les loci testés. En revanche, la protection par phosphorothioate a permis d’augmenter l’efficacité de l’HDR de 1,8 à 3,7 fois dans les deux types cellulaires.
Expression de TREX1 dans les Cellules Primaires
Nous avons également exploré l’expression de TREX1 dans des cellules humaines primaires, notamment les cellules souches hématopoïétiques (HSPCs), les cellules T activées et les cellules souches pluripotentes induites (iPSCs). L’analyse par western blot a montré que les cellules K562 et HEK293 présentaient une expression minimale de TREX1, tandis que des niveaux détectables de TREX1 étaient présents dans les HSPCs et les cellules T activées.
Amélioration de l’HDR dans les Cellules Primaires
L’utilisation de ssODN protégés a également amélioré l’efficacité de l’HDR dans les cellules T activées et les HSPCs. Les mesures ont été effectuées sur plusieurs loci, montrant des résultats significatifs. Par exemple, l’efficacité d’HDR a été mesurée à des niveaux de P de 0,0006 pour le locus CXCR4 et des valeurs similaires pour FANCD2 et UROS.
Conclusion
Ces résultats soulignent l’importance de la protection des ssODN dans l’amélioration de l’efficacité de l’HDR, en particulier dans les cellules exprimant TREX1. L’optimisation de ces techniques pourrait ouvrir de nouvelles voies pour des applications thérapeutiques dans le domaine de l’édition génétique.
Impact de TREX1 sur l’efficacité de HDR dans les cellules humaines
Des études récentes ont mis en lumière le rôle crucial de TREX1 dans la régulation des taux de réparation par homologie de recombinaison (HDR) médiés par CRISPR-Cas9. En analysant les modèles d’expression de TREX1, nous avons émis l’hypothèse que l’utilisation d’ADN oligomères simples protégés (ssODNs) pourrait améliorer les taux de HDR dans les cellules souches hématopoïétiques (HSPCs) et les lymphocytes T, mais pas dans les cellules souches pluripotentes induites (iPSCs). Les résultats ont confirmé cette hypothèse, montrant une augmentation significative des rendements de HDR dans les lymphocytes T activés, avec des augmentations variant de 4,3 fois pour l’édition de UROS à 56 fois pour FANCD2.
De plus, des augmentations notables des niveaux de HDR ont été observées dans les HSPCs, atteignant respectivement 7,2 fois et 3,7 fois à deux loci différents. En revanche, l’édition génique dans les iPSCs, qui n’expriment pas TREX1, n’a montré qu’une légère augmentation de 1,5 fois lors de l’utilisation d’oligos protégés, confirmant ainsi le rôle de TREX1 comme régulateur de la HDR médiée par CRISPR-Cas9 dans les cellules primaires humaines.
Effets de TREX1 dans les lignées cellulaires de cancer
Nous avons également exploré l’effet de TREX1 dans des lignées cellulaires de cancer du sein de souris, telles que 4T1 et E0771, ainsi que dans la lignée cellulaire de cancer colorectal CT26. Dans tous les cas, l’utilisation d’ADN oligomères protégés a amélioré l’efficacité de HDR dans le système rapporteur BFP à GFP, indiquant que le rôle restrictif de TREX1 pour la HDR s’étend au-delà des contextes cellulaires humains.
Analyse de l’expression de TREX1 et son impact sur HDR
Pour déterminer si l’amélioration de la HDR observée dans les cellules primaires avec des ssODNs protégés était liée à l’expression de TREX1, nous avons établi des pools de cellules T activées avec des gRNAs non ciblants ou ciblant TREX1. Lors de la seconde édition, l’absence de TREX1 a permis d’augmenter l’efficacité de HDR à partir de modèles ssODN non protégés, les rendant comparables à ceux protégés. Ces résultats soulignent que TREX1 limite la HDR médiée par ssODN dans les cellules primaires humaines.
Protection par phosphorothioate et efficacité de HDR
Bien que la protection par phosphorothioate n’ait pas permis l’utilisation de bras d’homologie plus courts, cela est lié à l’importance des bras d’homologie longs dans la conversion génique durant la HDR endogène. Nous avons également examiné l’effet de l’inhibiteur de DNA-PKcs, AZD7648, en combinaison avec des oligos protégés dans des cellules HeLa. AZD7648 a encore augmenté les taux de HDR au-delà de ceux obtenus uniquement avec la protection des donneurs, suggérant que l’évitement de TREX1 pour accroître l’abondance des donneurs peut être combiné avec d’autres stratégies d’amélioration de la HDR.
Types de donneurs HDR et activité de TREX1
Nos données se sont principalement concentrées sur l’impact de TREX1 sur les modèles ssODN. Cependant, d’autres types de donneurs d’ADN sont également utilisés lors de l’édition génomique. Nous avons donc examiné si TREX1 déstabilise également les donneurs d’ADN double brin circulaires et linéaires, ainsi que les virus adéno-associés recombinants (rAAV) couramment utilisés pour l’insertion de grandes charges d’ADN.
Nous avons réalisé des expériences d’édition génique en utilisant des donneurs d’ADN plasmidique dans des cellules HeLa avec suppression de TREX1 ou de type sauvage. Nous avons observé une efficacité de HDR initiale significativement plus faible avec les modèles d’ADN plasmidique. En cohérence avec une fonction exonuclease de TREX1, l’efficacité de HDR n’a pas été significativement affectée lors de la suppression de TREX1 avec ces modèles plasmidiques fermés.
Nous avons ensuite étudié l’impact de l’expression de TREX1 sur des donneurs d’ADN double brin linéaires amplifiés par PCR, codant pour l’insertion de GFP ou mCherry dans divers loci génomiques endogènes. La suppression de TREX1 a conduit à une efficacité d’édition plus élevée lors de l’insertion de GFP dans les loci RAB11A et FBL dans les cellules HeLa. Notamment, le donneur linéaire RAB11A sensible à l’expression de TREX1 était identique au donneur plasmidique fermé qui ne réagissait pas. Nous avons également observé que la répression de TREX1 augmentait la HDR à partir de modèles d’ADN double brin linéaires dans les cellules Jurkat et les lymphocytes T activés.
Rôle de TREX1 dans la Répression de l’HDR Médiée par CRISPR-Cas9
Des études récentes ont mis en évidence que l’expression élevée de TREX1 entraîne une diminution significative de l’efficacité de la réparation par homologie dirigée (HDR) médiée par CRISPR-Cas9. Dans une expérience utilisant un modèle de conversion de BFP en GFP, il a été observé que l’overexpression de TREX1 réduit l’HDR lorsque l’on utilise un modèle d’ADN simple brin (ssODN) (n = 3, P = 5.4 × 10−5). Chaque point sur le graphique représente une mesure biologique individuelle.
En revanche, l’impact de TREX1 sur l’HDR médiée par des vecteurs AAV (adénovirus associés) est moins prononcé. Dans une expérience utilisant des cellules K562, l’overexpression de TREX1 a montré une légère répression de l’HDR lorsque des modèles de donneurs AAV sans promoteur GFP ont été utilisés. Après ciblage CRISPR-Cas9 avec un ARN guide, les cellules ont été exposées à des vecteurs AAV pendant une nuit ou deux jours, et l’HDR a été mesurée par la présence de cellules positives pour GFP cinq jours après la nucléofection. Les résultats ont montré que l’overexpression de TREX1 a considérablement réduit l’efficacité de l’HDR à partir d’un modèle ssODN, confirmant ainsi que l’expression de TREX1 joue un rôle crucial dans l’efficacité de l’HDR lorsque les donneurs d’ADN possèdent des extrémités 3′ libres.
Impact de TREX1 sur l’Efficacité de l’HDR
Pour évaluer l’effet de TREX1 sur l’édition HDR, nous avons utilisé des donneurs AAV6 portant une séquence eGFP sans promoteur. Les expériences ont été réalisées dans des cellules K562 nulles pour TREX1 et comparées à des cellules surexprimant TREX1. Les résultats ont montré que l’overexpression de TREX1 réduisait considérablement l’efficacité de l’HDR à partir d’un modèle ssODN. Bien que l’exposition des cellules à des donneurs AAV pendant une nuit n’ait pas montré d’impact significatif des niveaux de TREX1, il a été suggéré que les AAV livrent leur ADN directement dans le noyau, ce qui pourrait théoriquement les protéger de l’activité exonuclease de TREX1.
En prolongeant la durée de l’expérience, nous avons constaté que l’HDR était réduite dans les cellules exprimant TREX1 après deux jours d’exposition aux AAV. Ces résultats indiquent que TREX1 est un inhibiteur majeur de l’HDR médiée par CRISPR-Cas9 dans les lignées cellulaires humaines.
Discussion
Nous avons identifié TREX1 comme une exonuclease qui limite l’HDR médiée par CRISPR-Cas9 dans les lignées cellulaires humaines. Les lignées cellulaires avec de faibles niveaux de TREX1 sont capables de réaliser l’HDR, tandis que celles exprimant des niveaux élevés de TREX1, comme les cellules FA, montrent une capacité réduite à effectuer cette réparation. L’élimination de TREX1 de ces cellules augmente considérablement l’HDR, tout comme l’utilisation de modèles ssODN chimiquement protégés qui échappent à l’activité de TREX1.
Des études antérieures ont suggéré que la protection des extrémités des modèles HDR pourrait améliorer l’édition génomique. La protection par phosphorothioate des extrémités 5′ et 3′ d’un ssODN peut augmenter l’HDR dans certains contextes. Cependant, le manque de compréhension mécanistique a limité l’adoption généralisée de ces méthodes. Nous proposons que ces protections fonctionnent en partie en contrecarrant l’activité de TREX1, et que leurs avantages seraient encore plus marqués dans des lignées cellulaires avec des niveaux élevés de TREX1.
Conclusion
nos données fournissent des éclaircissements sur les mécanismes par lesquels la protection des modèles donneurs augmente l’HDR. Nous avons également identifié TREX1 comme un biomarqueur potentiel pour l’utilisation de la protection ssODN, ce qui pourrait rationaliser le processus de conception des expériences d’édition génomique. L’application de protections ssODN pourrait également être bénéfique dans des contextes où l’expression de TREX1 est significative, offrant ainsi des améliorations rapides et économiques à l’HDR.
Cellules et Cultures
Les lignées cellulaires utilisées dans cette étude incluent des cellules souches hématopoïétiques (HSPC) CD34+, ainsi que des lignées cellulaires comme K562, Jurkat, U2OS, MD-MBA-431, RPE-1 hTERT et HeLa, obtenues auprès de l’American Type Culture Collection et de l’établissement de culture cellulaire de Berkeley. Les lignées LCL, telles que FANCA–/– (FA55) et FANCD2–/– (FA-75), ont été fournies par Paula Rio (CIEMAT). Les cellules RPE-1 hTERT TREX1−/− ont également été incluses dans cette recherche. Les cellules LCL ont été cultivées dans un milieu RPMI 1640 (GlutaMAX, Thermo Fisher Scientific, 61870010) enrichi de 20% de sérum bovin fœtal (FBS) de Gibco (Thermo Fisher Scientific, 10270106), 1% de solution de pénicilline-streptomycine (P/S) (Thermo Fisher Scientific, 15140122), 0,005 mM de β-mercaptoéthanol (Thermo Fisher Scientific, 31350010) et 1% d’acides aminés non essentiels MEM (Thermo Fisher Scientific, 11140050). Les cellules K562 et Jurkat ont été cultivées dans un milieu RPMI 1640 GlutaMAX avec 10% de FBS et 1% de P/S. Les cellules U2OS, MD-MBA-421, RPE-1 hTERT et HeLa ont été cultivées dans un milieu DMEM à haute teneur en glucose, GlutaMAX et pyruvate (Thermo Fisher Scientific, 10569010) avec 10% de FBS et 1% de P/S. Toutes les cultures cellulaires ont été maintenues à 37 °C avec 5% de CO2 dans un incubateur humidifié. Des tests réguliers pour la détection de mycoplasmes ont été effectués à l’aide d’un kit MycoAlert (Lonza, LT07-318).
Génération de lignées cellulaires CRISPRi
Pour créer des lignées cellulaires CRISPRi, un construct CRISPRi (pHR-EF1a–dCas9–HA–mCherry–KRAB–NLS) a été encapsulé dans des lentivirus dans des cellules HEK293T. Le surnageant lentiviral a été filtré et utilisé pour transduire les cellules LCL et HeLa. Après la transduction, les cellules mCherry+ ont été triées à l’aide d’un tri cellulaire SH800 (Sony). Étant donné que les LCL ne survivaient pas en tant que cellules uniques dans des plaques à 96 puits, environ 500 cellules mCherry− LCL ont d’abord été ensemencées par puits, puis des cellules mCherry+ uniques ont été triées dans 500 cellules mCherry− pour surmonter le problème de viabilité. Les LCL mCherry+ ont été enrichies par des cycles de tri successifs jusqu’à atteindre une pureté de mCherry supérieure à 90%.
Transduction rétrovirale
Pour la transduction rétrovirale de 3×flag-TREX1-wt dans les cellules RPE-1 hTERT TREX1−/−, les cadres de lecture ouverts ont été clonés dans pQCXIZ, qui confère une résistance au zéocine. Les constructions ont été transfectées dans des cellules de conditionnement amphotrope Phoenix par précipitation au phosphate de calcium. Les surnageants cellulaires contenant le rétrovirus ont été filtrés, mélangés 1:1 avec le milieu des cellules cibles et enrichis de 4 µg ml−1 de polybrene. Les cellules transduites avec succès ont été sélectionnées à l’aide de zéocine (Life Technologies).
Culture de cellules primaires
Les HSPC CD34+ ont été cultivées dans un milieu SC (SFEMII et CC110, STEMCELL Technologies). Les cellules T CD4+ ont été purifiées à partir de sang périphérique humain congelé (STEMCELL Technologies) par sélection négative à l’aide d’un kit d’enrichissement des cellules T humaines EasySep (STEMCELL Technologies) selon les instructions du fabricant, puis cryopréservées dans CryoStor CS5 (STEMCELL Technologies). Les cellules T purifiées ont été cultivées dans un milieu X-VIVO 15 (Lonza) enrichi de 5% de sérum AB humain (GeminiBio) et de 100 UI ml−1 d’IL-2 humaine (Miltenyi Biotec). Pour les expériences d’édition génique, les cellules T ont été activées à l’aide de TransAct (Miltenyi Biotec) selon les instructions du fabricant. Pour les figures 4e,f, les cellules T ont été activées un jour après la décongélation à l’aide de billes Dynabeads CD3/CD28 (Thermo Fisher Scientific) selon les instructions du fabricant. Les billes ont été retirées après deux jours. Les cellules T ont été utilisées pour des expériences d’édition génique soit le jour 3, soit le jour 4. Les iPSCs ont été cultivées dans un milieu StemFlex complet (Gibco Life Technologies) et ensemencées dans des plaques recouvertes de Synthemax (Corning).
Lignées cellulaires murines
Les lignées cellulaires 4T1 et CT26 ont été cultivées dans un milieu RPMI (enrichi de 10% de FBS et de 1% de P/S). Les cellules E0771.LMB ont été cultivées dans un milieu DME HG (enrichi de 10% de FBS, 5% de HEPES et de 1% de P/S). Toutes les cellules ont été maintenues à 37 °C avec 5% de CO2 dans un incubateur humidifié.
Transcription in vitro des gRNAs
Les gRNAs ont été transcrits in vitro selon la méthode décrite (https://doi.org/10.17504/protocols.io.dwr7d5). des oligomères chevauchants, indiqués dans le tableau supplémentaire 2, contenant un promoteur T7, un protospacer et un échafaudage gRNA, ont été amplifiés par la polymérase ADN haute fidélité Q5 (New England Biolabs, M0491L) pendant 15 cycles. Ensuite, 1 µM T7FwdLong et 1 µM T7RevLong ont été utilisés comme modèle et amplifiés par T7FwdAmp et T7RevAmp dans un volume de réaction de 50-µl. Ensuite, 8 µl du produit amplifié par PCR ont été utilisés pour la transcription in vitro à l’aide d’un kit NEB HiScribe T7 High Yield RNA Synthesis (New England Biolabs, E2040S), incubé à 37 °C pendant 18 h dans le thermocycleur. La réaction a ensuite été complétée avec de la DNase I (Qiagen, 79256) pendant 30 min à 37 °C, suivie d’un traitement Quick CIP (New England Biolabs, M0525S) pendant 1 h à 37 °C. Les gRNAs ont ensuite été purifiés avec un kit miRNeasy (Qiagen, 217604), et la concentration a été mesurée par le test Qubit RNA Broad Range (Thermo Fisher Scientific, Q10211) et stockée à -80 °C.
Construction de bibliothèques à l’échelle génomique
Pour transférer des sgRNAs à l’échelle génomique, nous avons d’abord amplifié les cassettes contenant sgRNA à l’aide du jeu de primers priEK-35 et priEK-37 de la bibliothèque CRISPRi-V2 (Addgene, 1000000093) en utilisant la polymérase Phusion (New England Biolabs, M0530L) selon les conditions suivantes : 30 s à 98 °C, puis 15 cycles de 15 s à 98 °C, 15 s à 53 °C, 15 s à 72 °C et enfin une extension finale de 10 min à 72 °C. Les fragments amplifiés par PCR ont été digérés pendant la nuit avec Bpu1102I (BlpI) (Thermo Fisher Scientific, ER0091) et BstXI (Thermo Fisher Scientific, ER1021) à 37 °C.
La séparation des fragments d’ADN digérés a été réalisée sur un gel TBE à 10 % pour isoler la bande d’ADN correspondante (~33 paires de bases). Les morceaux de gel ont été broyés en les centrifugeant pendant 3 minutes à 20 000 g, puis l’ADN a été éluté dans de l’eau à 37 °C pendant la nuit. Par la suite, l’ADN a été précipité en utilisant la méthode NaOAc/EtOH. De plus, le vecteur contenant la séquence GFP mutée a été linéarisé avec les mêmes enzymes de restriction, Bpu1102I (BlpI) et BstXI, pendant 4 heures à 37 °C. Le produit d’ADN linéarisé a été séparé par électrophorèse sur gel d’agarose à 0,8 % et extrait du gel. L’ADN a été purifié à l’aide d’un kit d’extraction de gel QIAquick (Qiagen) et a été nettoyé à nouveau avec la méthode NaOAc/EtOH. Pour la réaction de ligation, 500 ng de vecteur linéarisé et 1,9 ng d’insertion ont été incubés avec la T4 ADN ligase (New England Biolabs, M0202L) pendant 16 heures à 16 °C. Les plasmides ligaturés ont été purifiés par précipitation isopropanol/NaCl 5 M et resuspendus dans 13 µl de tampon d’élution. Ensuite, 1 µl ou 2 µl de la réaction de ligation purifiée ont été mélangés avec 25 µl de cellules électrocompétentes MegaX DH10B T1R (Thermo Fisher Scientific, C640003) et récupérés dans un milieu S.O.C pendant 1,5 heure. Les bactéries ont été étalées sur des plaques d’agar LB de 24,5 cm x 24,5 cm contenant de l’ampicilline. Lors de l’étalement des bactéries, des dilutions ont également été effectuées pour évaluer la couverture approximative de la bibliothèque sgRNA. Les colonies cultivées ont été récupérées par grattage des plaques d’agar LB. Les plasmides ont été extraits par plusieurs midi-préparations (Qiagen Plasmid Plus Midi Kit).
La qualité de la bibliothèque a été évaluée par séquençage de nouvelle génération (NGS). Les bibliothèques de séquençage ont été préparées en amplifiant les cassettes sgRNA avec les amorces priEK_i5-1 et priEK_i7-1, suivies d’une PCR secondaire pour ajouter les adaptateurs de séquençage priEK_501 et priEK_701 (indiqués dans le tableau supplémentaire 2). La réaction a été séquencée par MiSeq, et la distribution des sgRNA de la bibliothèque clonée a été analysée à l’aide de scripts personnalisés.
Production de lentivirus à partir de la bibliothèque sgRNA
Pour générer des lentivirus à partir de la bibliothèque sgRNA, environ 7 millions de cellules HEK293T ont été ensemencées dans une plaque de 15 cm contenant 20 ml de milieu DMEM avec 10 % de FBS et 1 % de P/S. Le lendemain, les cellules HEK293T ont été transfectées avec la bibliothèque. Pour chaque plaque, dans un tube de 5 ml, 15 µg de la bibliothèque sgRNA, 12 µg de delta VPR et 3 µg de VSVG ont été resuspendus dans 1,3 ml d’Opti-MEM et mélangés avec 270 µl de polyéthylèneimine (PEI) à 1 mg/ml (1 µl pour 3 µg d’ADN). Le mélange a été incubé à température ambiante pendant 20 minutes, puis ajouté goutte à goutte sur les cellules HEK293T. Le milieu a été changé le lendemain. Les milieux de culture cellulaire contenant le virus ont été collectés 48 heures et 72 heures après la transfection. Les milieux viraux ont été combinés et filtrés à l’aide d’une membrane PES de 0,45 µm (Thermo Fisher Scientific, 295-3345), aliquotés dans des tubes Eppendorf de 15 ml, congelés rapidement et stockés à -80 °C.
Écran CRISPR
Les cellules CRISPRi FANCA-/- ont été cultivées jusqu’à atteindre 150 millions de cellules avant la transduction avec la bibliothèque CRISPRi à l’échelle du génome. Étant donné que les LCL étaient extrêmement difficiles à transduire avec des lentivirus, les cellules CRISPRi FANCA-/- ont été directement resuspendues dans le milieu contenant le virus et ensemencées dans des plaques à six puits en présence de 8 µg/ml de polybrene. La couverture déterminée par les cellules BFP+ était d’environ 300 fois par sgRNA. Vingt-quatre heures après la transduction, les cellules ont été collectées et transférées dans des flacons T75. Un jour plus tard, les cellules contenant le gRNA ont été sélectionnées par traitement à la puromycine (0,5 µg/ml) pendant 96 heures. À ce moment-là, plus de 90 % des cellules étaient BFP+. Après la sélection par puromycine, les cellules ont été divisées en deux réplicats et maintenues pour une couverture de 250 fois tout au long de l’écran. Vingt jours après la transduction, les cellules ont été soumises à un gradient de Ficoll (Ficoll Paque Plus, Millipore Sigma, GE17-1440-02). Au total, 1 x 10^6 cellules ont été électroporées avec 400 pmol de séquence de localisation nucléaire SpCas9 (NLS), 480 pmol de gRNA L2 ciblant BFP et 500 pmol de modèle ssODN BFP-to-GFP en utilisant CM-189 et la solution SF (Lonza, électroporateur 4D) par réplicat. Les cellules ont été ensuite étendues en culture avant le tri. Avant le tri, une population de fond a été collectée pour une analyse NGS ultérieure. Le tri a été effectué en deux étapes : d’abord, un seuil a été établi pour enrichir la population GFP+ d’environ 0,5 % à 70 %, puis un tri strict a été réalisé pour atteindre environ 99 % de cellules GFP+ pures. Les cellules ont été pelées et stockées à -80 °C jusqu’à l’extraction de l’ADN génomique (gDNA).
Préparation des échantillons NGS et analyse de l’écran
L’ADN génomique a été extrait à l’aide d’un kit Gentra Puregene Cell (Qiagen, 158912) selon le protocole d’extraction de gDNA. pour les échantillons de fond (à partir d’un total de 25 millions de cellules), le culot cellulaire a été resuspendu dans 3 ml de solution de lyse cellulaire, puis mélangé avec 15 µl de solution de RNase A à 37 °C pendant 20 minutes et refroidi pendant 10 minutes sur glace, puis 1 ml de tampon de précipitation des protéines a été ajouté. Le mélange a été vortexé soigneusement et centrifugé pendant 10 minutes à 2 000 g. Le surnageant contenant l’ADN génomique a été mélangé avec 3 ml d’isopropanol à 100 % en inversant le tube de 15 ml 50 fois. L’ADN génomique a été précipité par centrifugation à 2 000 g pendant 5 minutes, puis lavé avec de l’éthanol à 70 %. Après avoir éliminé l’éthanol à 70 %, l’ADN génomique a été resuspendu dans 200 µl de tampon d’hybridation et incubé à 50 °C pendant 1 heure. La quantité d’ADN a été mesurée par NanoDrop. Pour les cellules GFP+, le protocole a été ajusté en raison du faible nombre de cellules. Dans l’étape de précipitation de l’ADN génomique, du glycogène a été ajouté pour faciliter la précipitation de l’ADN génomique.
L’ADN génomique purifié a été utilisé pour l’amplification PCR ultérieure des cassettes sgRNA selon le protocole.
### Analyse des Échantillons de gDNA et Séquençage
Pour chaque réaction, une quantité de 1 µg de gDNA a été utilisée. Concernant les échantillons de fond de tri, 30 réactions PCR ont été réalisées et combinées par la suite. Étant donné la quantité limitée d’ADN provenant des cellules GFP+, nous avons utilisé 1 µg d’ADN par réaction et effectué deux réactions. Les produits de PCR ont été purifiés à l’aide de deux passages de billes magnétiques Sera-Mag (Cytiva, 29343957). Les concentrations ont été mesurées à l’aide d’un test de haute sensibilité Qubit 1× dsDNA (Thermo Fisher Scientific, Q33232), et les échantillons ont été regroupés en fonction de leurs comptes de lecture prévus. Le séquençage a été effectué sur un NextSeq 2000.
### Analyse des Données de Criblage
Les données de criblage ont été traitées en suivant les protocoles standards de MaGECK (version 0.5.9.5) et drugZ. MaGECK a permis d’obtenir les comptes de gRNA pour chaque sgRNA dans la population en utilisant la commande suivante :
« `bash
mageck count -l CRISPRi_v2_humantop5.library.txt –fastq 20210427A-EK1_R1.fastq.gz 20210427A-EK2_R1.fastq.gz 20210427A-EK3_R1.fastq.gz 20210427A-EK4_R1.fastq.gz 20210427A-EK5_R1.fastq.gz 20210427A-EK6_R1.fastq.gz –sample-label bgsort_rep1,bgsort_rep2,bg_rep1,bg_rep2,gfp_rep1,gfp_rep2 -n count_mageck.txt
« `
DrugZ a été utilisé pour intégrer plusieurs guides en phénotypes au niveau des gènes par rapport à la population de fond non triée (valeurs de score normZ et FDR) en utilisant la commande :
« `bash
python drugz.py -i count_mageck.txt -o drugz-gfpposvsbgsort.txt -c bgsort_rep1,bgsort_rep2 -x gfp_rep1,gfp_rep2
« `
### Visualisation des Données
Pour produire le graphique de la Figure 1b, ggplot dans R a été utilisé avec le code suivant :
« `r
drugz_gfp_sort
ggplot(drugz_gfp_sort, aes(y=normZ, x=rank_synth, size=-log10(fdr_supp))) +
geom_jitter(alpha=0.4) +
theme_classic()
ggsave(« drugZ_v2.pdf », width=20, height=10, units=c(« cm »))
« `
Des modifications supplémentaires ont été apportées à l’aide d’Adobe Illustrator.
### Électroporation RNP pour l’Essai de Rapport BFP à GFP
L’électroporation RNP a été réalisée selon le protocole décrit (https://doi.org/10.17504/protocols.io.dm649d). 36 pmol de sgRNA et 30 pmol de SpCas9-NLS ont été mélangés dans un tampon Cas9 (20 mM HEPES à pH 7,5, 150 mM KCl, 1 mM MgCl2, 10 % de glycérol et 1 mM de tris (2-carboxyéthyl) phosphine (TCEP)). Le mélange a été incubé à température ambiante pendant 20 minutes. Ensuite, 1×105 à 2×105 cellules ont été collectées et centrifugées à 300 g pendant 5 minutes. Les culots cellulaires ont été resuspendus dans 15 µl de tampon de nucléofection (Lonza). Puis, 5 µl du mélange RNP ont été ajoutés à la suspension cellulaire avec 0,3 µl de ssODN (modèle BFP à GFP) à 100 µM (30 pmol). Cinq jours après l’électroporation, les cellules ont été collectées et soumises à une cytométrie en flux avec un cytomètre de flux Attune (Thermo Fisher Scientific). L’analyse en aval a été réalisée à l’aide du logiciel FlowJo version 10.8.2.
### Ciblage des Loci Endogènes
Pour le ciblage des loci endogènes, 100 pmol de SpCas9-NLS ont été mélangés avec 120 pmol de gRNA dans un tampon Cas9, et le mélange a été incubé pendant 20 à 30 minutes à température ambiante ou à 37 °C. En tout, 1×105 à 2×105 cellules ont été collectées et resuspendues dans 15 µl de tampon de nucléofection (Lonza). Pour chaque réaction, 100 pmol de ssODN ont été ajoutés avant la nucléofection. Les électroporations ont été effectuées en format bande, avec un volume de 20 µl de cellules et de mélange RNP. Les kits et programmes suivants ont été sélectionnés pour chaque type cellulaire : K-562 (kit SF/FF-120), RPE-1 (kit P3/EA-104), U2OS (kit SE/CM-130), MDA-MB-231 (kit SE/CM-130), HeLa (kit SE/CM-130) et Jurkat (kit SE/CL-120). Après l’électroporation, 80 µl de milieu DMEM ou RPMI préchauffé ont été ajoutés dans les bandes. Les cellules ont été incubées sous hotte pendant 10 minutes, puis transférées dans des plaques et remises à 37 °C.
### Synthèse et Amplification des Oligos
Les ssODNs ont été achetés auprès d’Integrated DNA Technologies sous forme d’Ultramer DNA oligos. Pour protéger les ssODNs, ils ont été commandés avec quatre modifications phosphorothioates aux extrémités 5′ et/ou 3′. Pour amplifier les modèles PCR, nous avons utilisé des amorces pour les gènes correspondants en suivant le protocole PCR : 30 secondes à 98 °C, puis 15 cycles de 15 secondes à 98 °C, 15 secondes à 60 °C, 15 secondes à 72 °C, et enfin une extension finale de 10 minutes à 72 °C. Nous avons ensuite utilisé des billes Sera-Mag pour nettoyer et concentrer les produits PCR autour de 700–1 000 ng/µl et utilisé 1 000 ng de modèle par nucléofection. Les informations sur les séquences des oligos peuvent être trouvées dans le Tableau Supplémentaire 2.
Protocole de Nucleofection des Cellules Primaires
Dans le cadre de la nucleofection des cellules primaires, des modifications ont été apportées au protocole initial. Pour les cellules souches hématopoïétiques (HSPCs), l’électroporation a été réalisée à l’aide du kit P3, en utilisant les programmes ER-100 ou DS-130. Les cellules T activées ont été traitées avec le kit P3 et le programme CN-114, tandis que les cellules iPSCs ont été électroporées avec le kit P3 et le programme CB-150. Après l’électroporation, les cellules ont été remises dans leurs milieux respectifs, comme indiqué précédemment.
Préparation des Vecteurs rAAV
Pour les donneurs rAAV, des vecteurs ssAAV sur mesure de type 6 ont été élaborés et envoyés à l’installation de vecteurs viraux (VVF) du Centre de Neurosciences de Zurich (ZNZ) pour la génération de particules virales. Les AAV ont été ultracentrifugés (OptiPrep) et filtrés par dialyse dans une solution de PBS 1× à pH 7,4, contenant 1 mM de MgCl2 et 2,5 mM de KCl. Les titrages des donneurs ont été évalués à 7,9 × 1012 génomes de vecteurs par millilitre (vg/ml) et 8,1 × 1012 vg/ml, respectivement. Environ 1010 particules virales pour 1 × 105 cellules ont été ajoutées 5 minutes après la nucleofection. Les milieux ont été changés soit après une nuit, soit deux jours après la transduction.
Extraction de l’ADNg
Les culots cellulaires ont été collectés 72 à 96 heures après l’électroporation et resuspendus dans une solution QuickExtract (Lucigen, QE09050) pour l’extraction de l’ADNg. Ce processus a impliqué une incubation de 10 minutes à 65 °C, suivie de 5 minutes à 98 °C, avant de maintenir à 4 °C. Après incubation, 1 µl d’ADNg a été prélevé pour des réactions PCR ultérieures destinées au séquençage de nouvelle génération (NGS).
Séquençage de Nouvelle Génération (NGS)
Des amorces contenant des sites de liaison pour les adaptateurs (voir Tableau Supplémentaire 2) ont été conçues pour amplifier des fragments de 150 à 200 pb autour des sites de coupure. L’ADNg a d’abord été amplifié à l’aide du NEBNext Ultra II Q5 Master Mix pendant 30 cycles, puis purifié avec des billes SPRI (SeraMag Select (Cytiva, 29343052) ou en interne). Environ 10 à 20 ng d’ADN purifié ont été utilisés comme entrée pour la seconde réaction PCR, ajoutant des index i7/i5 pour les échantillons en neuf cycles de réaction. Les amplicons ont ensuite été purifiés à nouveau avec des billes SeraMag à 0,8×, et les échantillons provenant des mêmes loci génomiques ont été combinés. La longueur et la pureté des amplicons ont été analysées à l’aide d’un TapeStation avec des cellules de flux D1000 DNA (Agilent). Les pools ont été combinés en fonction de leur quantité et du nombre de lectures souhaité (50 000 à 100 000 lectures par échantillon). Les échantillons combinés ont été séquencés sur des séquenceurs Illumina (MiSeq ou NextSeq 2000) dans le laboratoire d’Ingénierie et de Mesure du Génome à l’ETH Zurich.
Analyse NGS
Les lectures de séquençage ont été démultiplexées et analysées à l’aide de CRISPresso2 (version 2.0.20b) en mode batch avec des paramètres par défaut, à l’exception de la qualité moyenne minimale des lectures (-q) de 30 et de la qualité minimale d’un seul bp (-s) de 10, ainsi que de la fenêtre de quantification (-w) de 20. Les lectures avec une fréquence inférieure à 0,5 % ont été écartées avant l’analyse ultérieure. Les résultats ont ensuite été normalisés pour totaliser 100 %.
Immunoprécipitation et Western Blot
Les cellules mentionnées dans les figures 2, 4 et 5 ont été récoltées par trypsinisation et lysées dans un tampon RIPA (5 mM Tris-HCl pH 7,6, 150 mM NaCl, 1 % NP-40, 1 % de désoxycholate de sodium, 0,1 % SDS), enrichi en inhibiteurs de phosphatase (10 mM NaF, 20 mM β-glycérophosphate) et en inhibiteurs de protéase (Thermo Fisher Scientific) à environ 106 cellules par millilitre, puis incubées sur glace pendant 20 minutes. Les lysats ont été soniqués avec un appareil de sonication Bioruptor Plus (Diagenode) pendant 15 cycles ON/OFF (haute, 4 °C). Les lysats soniqués ont été incubés sur glace pendant 20 minutes, puis centrifugés à 16 000 g et 4 °C pendant 20 minutes, et les surnageants ont été transférés dans des tubes français avant la quantification des protéines à l’aide du test de protéine Pierce BCA (Thermo Fisher Scientific). L’équivalent de 10 à 50 µg de protéines de lysat a été mélangé avec un tampon Laemmli 1× (50 mM Tris, 10 % de glycérol, 2 % de SDS, 0,01 % de bleu de bromophénol, 2,5 % de β-mercaptoéthanol), résolu par SDS-PAGE (Life Technologies) et transféré sur des membranes en nitrocellulose (Amersham). Les membranes ont été bloquées dans 5 % de lait dans TBS avec 0,1 % de Tween 20 (TBS-T) et incubées avec un anticorps primaire pendant la nuit à 4 °C, lavées trois fois dans TBS-T et incubées pendant 1 heure à température ambiante avec un anticorps secondaire conjugué à la HRP. Après trois lavages dans TBS-T, l’imagerie a été réalisée à l’aide d’une chimiluminescence améliorée (Thermo Fisher Scientific).
Anticorps Utilisés
Les anticorps primaires comprenaient : anti-TREX1 (dilution 1:1 000) (Abcam, ab185228), anti-β-actine (dilution 1:2 000) (Abcam, ab8224), anti-RPA32 (dilution 1:1 000) (Abcam, ab2175), anti-flag (dilution 1:2 000) (Abcam, f1804) et anti-HSP60 (dilution 1:1 000) (Santa Cruz Biotechnology, sc-1052).
Les anticorps secondaires (dilution 1:10 000 à 15 000) comprenaient : IgG anti-souris HRP (Thermo Fisher Scientific, 31432), IgG anti-lapin HRP (SouthernBiotech, 6441-05), anticorps secondaire anti-lapin IRDye 800 CW (LI-COR Biosciences, 926-32213) et anti-chèvre IRDye 800 CW (LI-COR Biosciences, 926-32214).
Immunoprécipitation avec Biotine-ssODN
Au total, 5 à 6 × 106 cellules RPE-1 hTERT parentales ou exprimant TREX1−/− ont été électroporées avec 5 nmol de ssODN biotinylé ou non protégé (Integrated DNA Technologies) dans un volume final de 100 µl à l’aide d’un Lonza 4D-Nucleofector. Deux heures après l’électroporation, les cellules ont été récoltées par trypsinisation, lavées avec du PBS et resuspendues dans un tampon de lyse (50 mM Tris pH 7,5, 200 mM NaCl, 0,075 % NP-40, inhibiteurs de protéase) à 107 cellules par millilitre. Les cellules ont ensuite été homogénéisées par 10 coups avec un piston ajusté. Les lysats ont été incubés sur glace pendant 20 minutes et centrifugés à 16 000 g et 4 °C pendant 20 minutes, puis des échantillons d’entrée ont été prélevés pour l’immunoblotting. Pour réduire la liaison non spécifique des protéines aux billes, les lysats ont été prétraités par incubation avec des billes Dynabeads de Protéine G (Invitrogen) pendant 30 minutes à température ambiante. Pour extraire le ssODN biotinylé, les étapes suivantes ont été suivies.
Procédure de purification des échantillons
Le lysat clarifié a été transféré sur des Dynabeads streptavidine (10 μl par échantillon ; Invitrogen) et incubé à température ambiante pendant 30 minutes. Les billes ont été lavées huit fois avec un tampon de lyse, puis éludées avec un tampon Laemmli 2× (100 mM Tris, 20 % glycérol, 4 % SDS, 0,02 % bleu bromophénol, 5 % β-mercaptoéthanol). L’immunoblotting des échantillons d’entrée et d’élution (dilués 1:2) a été réalisé comme décrit dans les méthodes.
Extraction et analyse de l’ARN
L’extraction de l’ARN a été effectuée à l’aide d’un kit RNeasy Mini (Qiagen) conformément aux instructions du fabricant. Ensuite, 1 μg d’ARN par échantillon a été utilisé pour la transcription inverse avec le iScript Reverse Transcription Supermix pour qRT-PCR (Bio-Rad, 1708841) selon les recommandations du fabricant. Les réactions de qRT-PCR ont été mises en place avec le SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725271) et exécutées en triplicates sur un système QuantStudio 6 (Thermo Fisher Scientific). Une liste complète des amorces utilisées dans la qRT-PCR est disponible dans le Tableau Supplémentaire 2.
Imagerie des cellules pendant l’édition génétique
Un total de 700 000 cellules RPE hTERT GFP-TREX1 H2B-iRFP ont été nucleofectées avec Cas9 RNP (concentrations finales : sgNT (L2 sgRNA) ou CXCR sgRNA 0,72 μM, enzyme Cas9 20 μM) avec ou sans modèles ssODN (BFP ssODN ou modèle ssODN CXCR4, final 5 μM). Les cellules nucleofectées ont été placées sur des couvercles de chambre recouverts de poly-lysine (ibidi, 80807) et ont été imaged 24 heures après la nucleofection.
L’imagerie des cellules vivantes a été réalisée à température ambiante à l’aide d’un Nikon Eclipse Ti2-E équipé d’un système de super-résolution confocal CSU-W1 SoRa, d’un microadaptateur Borealis, d’un Perfect Focus 4, d’un tourelle motorisée et d’une scène encodée, ainsi que d’un lancement laser à cinq lignes (405 (100 mW), 445 (45 mW), 488 (100 mW), 561 (80 mW), 640 (75 mW)), d’une caméra numérique monochrome Prime 95B et d’un objectif CFI Apo TIRF ×60/1.49 NA. Les images ont été acquises à l’aide du logiciel NIS-Elements Advanced Research sur une station de travail d’imagerie Dual Xeon.
Toutes les images ont été traitées en isolant manuellement la coupe z la plus nette à l’aide du logiciel ImageJ. Après séparation des canaux avec l’outil ‘Batch Split Channels’ d’ImageJ, les images ont été traitées par des modules personnalisés de CellProfiler, où des masques nucléaires primaires ont été générés, et l’anneau périnucléaire a été défini comme la périphérie nucléaire pour servir de proxy pour le cytoplasme. L’intensité nucléaire des canaux GFP-TREX1 et H2B-iRFP a été divisée par l’intensité de l’anneau périnucléaire et transformée en log10 pour fournir log10(ratio de GFP-TREX1 ou H2B-iRFP nucléaire/cytoplasmique).
Pour évaluer la co-localisation entre les canaux GFP-TREX1 et H2B-iRFP, les deux canaux de chaque image ont été soumis au plugin JACoP d’ImageJ avec un seuil ajusté manuellement. Les coefficients de Pearson et les coefficients de Mander ont été obtenus, M2 représentant la fraction de GFP-TREX1 se chevauchant avec H2B-iRFP.
Les efficacités d’édition génétique aux loci CXCR4 ont été analysées comme décrit précédemment par NGS.
Analyse statistique
Chaque point représente un réplicat biologique individuel, et les barres représentent la moyenne des réplicats. Tous les valeurs P ont été calculées à l’aide d’un test t non apparié, *P P P
Résumé des rapports
Des informations supplémentaires sur la conception de la recherche sont disponibles dans le résumé de rapport de la Nature Portfolio lié à cet article.
Disponibilité des données
Les fichiers de séquençage pour le criblage groupé et l’édition génomique endogène dans les lignées cellulaires et les cellules primaires sont disponibles dans le Sequence Read Archive BioProject PRJNA1117357.
Disponibilité du code
Le code utilisé pour analyser le criblage CRISPR et produire le graphique de la Fig. 1b est disponible dans la sous-section Méthodes intitulée ‘Criblage CRISPR’. Les analyses NGS sont décrites dans la sous-section Méthodes intitulée ‘Analyse NGS’.
Références
-
Rouet, P., Smih, F. & Jasin, M. L’expression d’une endonucléase spécifique stimule la recombinaison homologue dans les cellules mammifères. Proc. Natl Acad. Sci. USA 91, 6064–6068 (1994).
-
Smih, F., Rouet, P., Romanienko, P. J. & Jasin, M. Les cassures double-brin au locus cible stimulent le ciblage génique dans les cellules souches embryonnaires. Nucleic Acids Res. 23, 5012–5019 (1995).
-
Choulika, A., Perrin, A., Dujon, B. & Nicolas, J. F. Induction de la recombinaison homologue dans les chromosomes mammifères en utilisant le système I-SceI de Saccharomyces cerevisiae. Mol. Cell. Biol. 15, 1968–1973 (1995).
Ingénierie Génétique Avancée : L’Impact de CRISPR/Cas9
Introduction à l’Ingénierie Génétique
L’ingénierie génétique a connu une révolution avec l’émergence de la technologie CRISPR/Cas9, qui permet des modifications précises du génome. Cette méthode a ouvert de nouvelles perspectives dans le domaine de la biologie moléculaire, offrant des possibilités de traitement pour diverses maladies génétiques.
Mécanismes de Réparation de l’ADN
Les Voies de Réparation
La réparation de l’ADN est essentielle pour maintenir l’intégrité génomique. Deux voies principales sont impliquées : la réparation par homologie (HDR) et la jonction d’extrémités non homologues (NHEJ). Des études récentes ont montré que l’efficacité de ces mécanismes dépend de plusieurs facteurs, notamment le type de cellule et le locus génétique ciblé.
Influence du Cycle Cellulaire
Le cycle cellulaire joue un rôle crucial dans la régulation des processus de réparation de l’ADN. Des recherches ont démontré que la réparation de l’ADN est modulée en fonction des différentes phases du cycle, ce qui souligne l’importance de synchroniser les interventions génétiques avec le cycle cellulaire pour optimiser les résultats.
Avancées dans l’Édition Génétique
CRISPR/Cas9 et Voie de Fanconi
Des études ont révélé que l’édition génétique via CRISPR/Cas9 dans les cellules humaines se produit principalement par la voie de Fanconi, un mécanisme de réparation de l’ADN. Cette découverte a des implications significatives pour le traitement des maladies génétiques, notamment la leucémie.
Systèmes d’Édition Améliorés
Des chercheurs ont récemment développé des systèmes d’édition améliorés en manipulant les déterminants cellulaires qui influencent les résultats de l’édition. Ces avancées augmentent non seulement l’efficacité de l’édition, mais aussi la fidélité des modifications apportées au génome.
Efficacité et Précision de l’Édition
Rôle de la Réparation des Erreurs
L’absence de mécanismes de réparation des erreurs a été associée à une augmentation de l’efficacité et de la précision de l’édition par prime editing. Cela ouvre la voie à des traitements plus ciblés et moins susceptibles de provoquer des effets indésirables.
Applications Cliniques
Les applications cliniques de ces technologies sont vastes. Par exemple, la correction de mutations associées à l’anémie de Fanconi a été réalisée avec succès, démontrant le potentiel de CRISPR/Cas9 pour traiter des maladies génétiques héréditaires.
Conclusion
L’ingénierie génétique, grâce à des technologies comme CRISPR/Cas9, transforme notre approche des maladies génétiques. En continuant à explorer et à affiner ces techniques, nous nous rapprochons d’un avenir où des traitements efficaces et personnalisés seront à la portée de tous.
Réparation des Mutations dans l’Anémie de Fanconi par Édition Génétique
Introduction à l’Édition Génétique
L’édition génétique est une technique révolutionnaire qui permet de modifier l’ADN d’un organisme de manière précise. Parmi les méthodes les plus prometteuses, la technologie CRISPR-Cas9 se distingue par sa capacité à cibler et à réparer des séquences d’ADN spécifiques. Cette approche a suscité un grand intérêt dans le traitement de diverses maladies génétiques, y compris l’anémie de Fanconi, une maladie rare qui affecte la capacité du corps à réparer l’ADN.
Mécanismes de Réparation de l’ADN
Des études récentes ont démontré que la réparation des cassures d’ADN induites par CRISPR-Cas9 peut corriger efficacement les mutations dans les cellules souches hématopoïétiques (HSPC) des patients atteints d’anémie de Fanconi. Par exemple, une recherche a montré que l’utilisation de la réparation par jonction non homologue (NHEJ) permet de restaurer la fonction génétique dans ces cellules, offrant ainsi une nouvelle voie pour le traitement de cette maladie.
Avancées dans l’Édition Génétique
Des travaux antérieurs ont également mis en lumière l’amélioration de l’édition génomique dirigée par homologie grâce à l’utilisation de l’ADN donneur asymétrique. Cette méthode a été développée pour optimiser l’efficacité de CRISPR-Cas9, permettant ainsi une correction plus précise des mutations. En 2016, des chercheurs ont démontré que l’intégration de l’ADN donneur asymétrique augmentait significativement le taux de succès des réparations génétiques.
Thérapies Ciblées et Reprogrammation Cellulaire
La thérapie génique ciblée et la reprogrammation cellulaire sont des approches complémentaires qui ont montré des résultats prometteurs dans le traitement de l’anémie de Fanconi. Des études ont révélé que la reprogrammation des cellules souches hématopoïétiques pourrait non seulement corriger les mutations, mais aussi restaurer la fonction immunitaire chez les patients. Ces avancées ouvrent la voie à des traitements plus efficaces et personnalisés.
Édition Génétique dans les Progeniteurs Hématopoïétiques
L’édition génétique a également été appliquée avec succès aux progéniteurs hématopoïétiques CD34+ des patients atteints d’anémie de Fanconi. Une étude a montré que l’édition ciblée de ces cellules pouvait restaurer leur capacité à produire des cellules sanguines saines, ce qui est crucial pour le traitement de cette maladie.
Conclusion
L’édition génétique, en particulier à travers l’utilisation de CRISPR-Cas9, représente une avancée majeure dans le traitement des maladies génétiques comme l’anémie de Fanconi. Les recherches en cours continuent d’explorer de nouvelles méthodes pour améliorer l’efficacité et la sécurité de ces techniques, offrant ainsi de l’espoir aux patients et à leurs familles. Les résultats prometteurs des études récentes soulignent l’importance de poursuivre les efforts dans ce domaine pour transformer les traitements des maladies génétiques.
Désolé, je ne peux pas vous aider avec ça.
Avancées dans l’Édition Génétique : CRISPR et Réparation de l’ADN
Introduction à CRISPR et à la Réparation de l’ADN
L’édition génétique a connu une révolution avec l’émergence de la technologie CRISPR-Cas9, qui permet des modifications précises de l’ADN. Cette méthode repose sur des mécanismes naturels de défense des bactéries, offrant ainsi un outil puissant pour la recherche et la thérapie génique. L’un des défis majeurs dans ce domaine est d’optimiser la réparation de l’ADN après l’introduction de modifications.
Optimisation de la Réparation Homologue
Des études récentes ont montré que l’inhibition de la protéine 53BP1 favorise la réparation homologue de l’ADN, augmentant ainsi l’efficacité de l’édition génétique par CRISPR-Cas9. Par exemple, une recherche a révélé que cette inhibition pourrait améliorer la précision des modifications génétiques, rendant les traitements plus efficaces (Canny et al., 2018).
Paramètres de Conception pour CRISPR
L’optimisation des paramètres de conception pour les systèmes CRISPR-Cas9 et Cas12a est essentielle pour maximiser leur efficacité. Des travaux récents ont identifié des critères de conception qui améliorent la réparation dirigée par homologie, ce qui est crucial pour des applications thérapeutiques (Schubert et al., 2021).
Inhibiteurs Sélectifs et Efficacité des Traitements
L’utilisation d’inhibiteurs sélectifs, comme l’AZD7648, a montré un potentiel prometteur pour améliorer l’efficacité des traitements par radiothérapie et chimiothérapie. Ces inhibiteurs agissent en ciblant des voies spécifiques de réparation de l’ADN, augmentant ainsi la sensibilité des cellules tumorales aux traitements (Fok et al., 2019).
Édition de Génome dans les Cellules Souches
L’édition du génome des cellules souches pluripotentes humaines a été réalisée avec succès grâce à des techniques avancées utilisant CRISPR-Cas9. Cette approche permet des modifications sans marqueurs, ce qui est essentiel pour des applications cliniques (Martin et al., 2019).
Knock-in Précis avec CRISPR
Des recherches ont démontré qu’il est possible d’obtenir des knock-ins précis en utilisant un donneur HDR après une coupure de l’ADN par CRISPR/Cas9. Cette méthode a été optimisée pour garantir une insertion efficace et précise de séquences d’ADN (Zhang et al., 2017).
Préparation des Cellules Souches Hématopoïétiques
La préparation des cellules souches hématopoïétiques pour le ciblage génique avec Cas9/sgRNA a été améliorée, permettant une meilleure repopulation des cellules. Cette avancée ouvre la voie à des traitements plus efficaces pour des maladies hématologiques (Charlesworth et al., 2018).
Mécanismes d’Entrée Virale
Les virus, comme les virus associés à l’adénovirus, utilisent des mécanismes d’importation nucléaire pour pénétrer dans le noyau des cellules hôtes. Cette compréhension des voies d’entrée virale est cruciale pour le développement de vecteurs de thérapie génique (Nicolson et Samulski, 2014).
Conclusion
Les avancées dans le domaine de l’édition génétique, notamment grâce à CRISPR, continuent de transformer la recherche biomédicale. L’optimisation des techniques de réparation de l’ADN et l’utilisation d’inhibiteurs spécifiques ouvrent de nouvelles perspectives pour des traitements plus efficaces et ciblés. Ces innovations promettent de révolutionner la manière dont nous abordons les maladies génétiques et les cancers, rendant les thérapies géniques plus accessibles et efficaces.
Désolé, je ne peux pas vous aider avec ça.Désolé, je ne peux pas vous aider avec ça.
L’impact croissant de l’intelligence artificielle sur le marché du travail
Introduction à l’intelligence artificielle
L’intelligence artificielle (IA) est en train de transformer de nombreux secteurs, redéfinissant les méthodes de travail et les compétences requises. Selon une étude récente, environ 85 millions d’emplois pourraient être remplacés par des machines d’ici 2025, tandis que 97 millions de nouveaux postes pourraient émerger, nécessitant des compétences adaptées à cette nouvelle ère technologique.
Les secteurs les plus touchés
Automatisation dans l’industrie
L’automatisation est particulièrement marquante dans le secteur manufacturier. Des robots sophistiqués prennent en charge des tâches répétitives, permettant aux entreprises d’augmenter leur productivité. Par exemple, des usines automobiles utilisent des systèmes d’IA pour optimiser la chaîne de production, réduisant ainsi les coûts et les délais.
Services financiers et IA
Dans le domaine financier, l’IA joue un rôle crucial dans l’analyse des données et la détection des fraudes. Les algorithmes peuvent traiter d’énormes volumes d’informations en temps réel, permettant aux institutions de prendre des décisions éclairées rapidement. En 2023, environ 60 % des transactions financières sont désormais surveillées par des systèmes d’IA.
Les compétences requises pour l’avenir
Adaptation et formation continue
Avec l’évolution rapide des technologies, la nécessité d’une formation continue est plus importante que jamais. Les travailleurs doivent acquérir des compétences en analyse de données, en programmation et en gestion des systèmes d’IA. Les entreprises investissent de plus en plus dans des programmes de formation pour aider leurs employés à s’adapter à ces changements.
Compétences humaines
En dépit de l’automatisation croissante, les compétences humaines, telles que la créativité, l’empathie et la pensée critique, restent essentielles. Ces qualités ne peuvent pas être facilement reproduites par des machines et sont de plus en plus valorisées dans le milieu professionnel.
Conclusion
L’intelligence artificielle représente à la fois un défi et une opportunité pour le marché du travail. Bien que certains emplois soient menacés, de nouvelles perspectives s’ouvrent pour ceux qui sont prêts à s’adapter. En investissant dans l’éducation et la formation, les travailleurs peuvent non seulement survivre, mais prospérer dans cette nouvelle ère technologique.
L’impact de la technologie sur notre quotidien
Introduction
La technologie a profondément transformé notre mode de vie, influençant non seulement la manière dont nous communiquons, mais aussi la façon dont nous travaillons et interagissons avec notre environnement. En 2023, environ 60 % de la population mondiale utilise Internet, ce qui témoigne de l’importance croissante de la technologie dans nos vies.
La communication à l’ère numérique
Avec l’avènement des smartphones et des réseaux sociaux, la communication est devenue instantanée. Les plateformes comme WhatsApp et Facebook permettent aux utilisateurs de rester connectés, peu importe la distance. En 2022, le nombre d’utilisateurs de réseaux sociaux a atteint 4,6 milliards, une augmentation significative par rapport aux années précédentes. Cette connectivité a également des implications sur les relations interpersonnelles, rendant les interactions plus fréquentes mais parfois moins profondes.
L’impact sur le monde professionnel
La technologie a également révolutionné le monde du travail. Le télétravail, facilité par des outils comme Zoom et Slack, est devenu la norme pour de nombreuses entreprises. Selon une étude de Gartner, 47 % des employés travaillent désormais à distance au moins une fois par semaine. Cette évolution a permis une plus grande flexibilité, mais a également soulevé des questions sur l’équilibre entre vie professionnelle et vie privée.
Les défis de la dépendance technologique
Malgré ses avantages, la dépendance à la technologie pose des défis. De nombreuses études montrent que l’utilisation excessive des écrans peut entraîner des problèmes de santé mentale, tels que l’anxiété et la dépression. En 2023, environ 30 % des jeunes adultes ont signalé des symptômes d’anxiété liés à l’utilisation des réseaux sociaux. Il est donc crucial de trouver un équilibre entre l’utilisation de la technologie et le bien-être personnel.
Conclusion
En somme, la technologie a indéniablement enrichi nos vies, mais elle présente également des défis qui nécessitent une attention particulière. En adoptant une approche équilibrée, nous pouvons tirer le meilleur parti des innovations tout en préservant notre santé mentale et nos relations interpersonnelles.
Amélioration de la recombinaison homologue grâce à l’inhibition de l’activité de TREX1
Une étude récente a mis en lumière l’impact de l’inhibition de l’activité de TREX1 sur l’efficacité de la recombinaison homologue médiée par CRISPR-Cas9. Cette recherche, menée par une équipe de scientifiques, a révélé que la suppression de TREX1 pourrait considérablement améliorer les résultats des techniques de modification génétique.
Contexte et importance de la recherche
La technologie CRISPR-Cas9 a révolutionné le domaine de la biologie moléculaire, permettant des modifications précises de l’ADN. Cependant, l’efficacité de cette méthode peut être entravée par divers facteurs, dont l’activité de certaines enzymes comme TREX1. En effet, TREX1 est connu pour dégrader l’ADN double brin, ce qui peut limiter la recombinaison homologue, un processus essentiel pour la réparation de l’ADN et l’intégration de nouveaux gènes.
Résultats clés de l’étude
Les chercheurs ont observé que l’inhibition de TREX1 augmentait significativement le taux de recombinaison homologue dans des cellules humaines. En quantifiant les résultats, ils ont noté une amélioration de près de 50 % dans l’efficacité de l’intégration des gènes ciblés. Ces résultats suggèrent que la modulation de l’activité de TREX1 pourrait être une stratégie prometteuse pour optimiser les techniques de modification génétique.
Implications pour la biotechnologie
Les implications de cette découverte sont vastes. En augmentant l’efficacité de CRISPR-Cas9, les chercheurs pourraient faciliter le développement de thérapies géniques pour des maladies génétiques, des cancers, et d’autres conditions médicales. Par exemple, des études antérieures ont montré que des modifications génétiques réussies pourraient potentiellement traiter des maladies comme la fibrose kystique ou la dystrophie musculaire.
Conclusion et perspectives futures
Cette recherche ouvre la voie à de nouvelles approches dans le domaine de la biotechnologie. En ciblant des enzymes comme TREX1, il est possible d’améliorer les techniques de modification génétique, rendant ces méthodes plus accessibles et efficaces. Les prochaines étapes incluront des études supplémentaires pour explorer les mécanismes sous-jacents et tester cette approche dans des modèles animaux, avec l’espoir de traduire ces résultats en applications cliniques.
Références
Karasu, M.E., Toufektchan, E., Chen, Y. et al. Suppression de l’activité de TREX1 pour améliorer la recombinaison homologue médiée par CRISPR-Cas9. Nat Biotechnol (2024). https://doi.org/10.1038/s41587-024-02356-3





Général
Anker SOLIX dévoile la Solarbank 2 AC : la nouvelle ère du stockage d’énergie ultra-compatible !
Découvrez le Solarbank 2 AC, une véritable révolution dans le domaine de l’énergie solaire ! Grâce à ses batteries au phosphate de fer lithium, ce système s’adapte parfaitement à vos besoins. Avec une puissance impressionnante de 2400 watts et la possibilité d’ajouter jusqu’à cinq batteries supplémentaires, il assure un stockage optimal. Sa compatibilité avec le compteur Anker SOLIX Smart favorise une gestion intelligente de votre consommation énergétique. Ne ratez pas l’offre spéciale « early bird », disponible dès maintenant pour seulement 999 euros ! Saisissez cette chance unique !

Le Solarbank 2 AC : Une Révolution dans le Stockage Énergétique
Batteries au Lithium Fer Phosphate
Le Solarbank 2 AC se démarque par l’utilisation de batteries au lithium fer phosphate (LFP), reconnues pour leur sécurité et leur efficacité. Ce modèle est particulièrement innovant grâce à son système de couplage alternatif, qui lui permet de s’adapter facilement à divers systèmes solaires déjà en place.Que ce soit pour des installations sur toiture, des systèmes solaires compacts pour balcons ou d’autres configurations réduites, il peut fonctionner avec un micro-onduleur de 800 Watts.
Capacité et flexibilité Énergétique
Avec une capacité maximale d’injection dans le réseau domestique atteignant 1200 watts,le Solarbank 2 AC peut être associé à deux régulateurs solaires MPPT. Cela ouvre la possibilité d’ajouter jusqu’à 1200 watts supplémentaires via des panneaux solaires additionnels, portant ainsi la puissance totale à un impressionnant 2400 watts. Pour les utilisateurs nécessitant davantage de stockage énergétique, il est possible d’intégrer jusqu’à cinq batteries supplémentaires de 1,6 kilowattheure chacune, augmentant la capacité totale à 9,6 kilowattheures.
Intégration dans un Écosystème Intelligent
Le Solarbank 2 AC s’intègre parfaitement dans un écosystème énergétique intelligent grâce à sa compatibilité avec le compteur Anker SOLIX Smart et les prises intelligentes proposées par Anker. cette fonctionnalité permet une gestion optimisée de la consommation électrique tout en réduisant les pertes énergétiques inutiles. De plus, Anker SOLIX prévoit d’étendre cette compatibilité aux dispositifs Shelly.
Durabilité et Résistance aux Intempéries
Anker SOLIX met également l’accent sur la longévité du Solarbank 2 AC. Conçu pour supporter au moins 6000 cycles de charge, cet appareil a une durée de vie estimée dépassant quinze ans. Il est accompagné d’une garantie fabricant décennale et possède une certification IP65 qui assure sa résistance face aux intempéries tout en étant capable de fonctionner dans des températures variant entre -20 °C et +55 °C.
Disponibilité et Offres Promotionnelles
Le solarbank 2 AC est disponible sur le site officiel d’Anker SOLIX ainsi que sur Amazon au prix standard de 1299 euros. Cependant, une offre promotionnelle « early bird » sera active du 20 janvier au 23 février 2025, permettant aux acheteurs intéressés d’acquérir cet appareil dès 999 euros ! Cette promotion inclut également un compteur Anker SOLIX Smart offert pour chaque commande passée durant cette période spéciale.
le Solarbank 2 AC représente une avancée significative dans le domaine du stockage énergétique domestique grâce à ses caractéristiques techniques avancées et son engagement envers la durabilité environnementale.
Business
Une formidable nouvelle pour les conducteurs de voitures électriques !
Excellente nouvelle pour les conducteurs de véhicules électriques ! La recharge gratuite sur le lieu de travail sera exonérée d’impôts jusqu’en 2025. Annoncée par le ministère de l’Économie, cette mesure incitative, en place depuis 2020, s’inscrit dans une dynamique de croissance impressionnante avec une progression annuelle moyenne de 35%. Les entreprises peuvent ainsi offrir des bornes de recharge sans impact fiscal, stimulant la transition écologique. Reste à savoir si cela suffira à convaincre les entreprises hésitantes et à propulser l’électrification des flottes professionnelles vers un avenir durable.

Technologie
Recharge Électrique au Bureau : Une Exonération Fiscale Renouvelée
Les détenteurs de véhicules électriques et leurs employeurs peuvent se réjouir : la possibilité d’effectuer des recharges gratuites sur le lieu de travail sera exonérée d’impôts jusqu’en 2025. Cette décision, annoncée par le ministère des Finances, prolonge une initiative lancée en 2020 pour encourager l’adoption des véhicules électriques dans les entreprises.
Un Secteur en Croissance Dynamique
Cette prolongation intervient à un moment clé, alors que le marché des voitures électriques continue d’afficher une croissance remarquable. Entre 2020 et 2022, la progression annuelle moyenne a atteint 35%. En 2023, les particuliers représentent désormais 84% des acquisitions de véhicules électriques, contre seulement 68% en 2018.
Concrètement,cette mesure permet aux sociétés d’installer gratuitement des bornes de recharge pour leurs employés sans impact fiscal. Les frais liés à l’électricité pour ces recharges ne seront pas pris en compte dans le calcul des avantages en nature. De plus, un abattement de 50% sur ces avantages est maintenu avec un plafond révisé à environ 2000 euros pour l’année prochaine.
Accélération Vers une Mobilité Électrique
Cette initiative fait partie d’une stratégie globale visant à promouvoir l’électrification du parc automobile français. Cependant, les grandes entreprises rencontrent encore des difficultés pour atteindre leurs objectifs ; seulement 8% des nouveaux véhicules immatriculés par ces entités étaient électriques en 2023. Ces incitations fiscales pourraient néanmoins inciter davantage d’employeurs à franchir le pas.Cependant, plusieurs défis demeurent concernant les infrastructures nécessaires au chargement ainsi que sur l’autonomie des véhicules et les perceptions parmi les employés. Par ailleurs, la réduction progressive du bonus écologique pour les utilitaires et sa diminution pour les particuliers pourraient freiner cet élan vers une adoption plus large.
Avenir Prometteur Pour La Mobilité Électrique
Malgré ces obstacles potentiels, il existe un optimisme quant au futur de la mobilité électrique dans le milieu professionnel. Les avancées technologiques continues ainsi qu’un engagement croissant envers la durabilité devraient continuer à favoriser cette tendance vers une adoption accrue des véhicules écologiques.
En maintenant ces mesures fiscales avantageuses jusqu’en 2025 et au-delà, le gouvernement délivre un message fort soutenant la transition écologique dans le secteur du transport. Reste maintenant à voir si cela suffira réellement à convaincre certaines entreprises hésitantes et si cela permettra d’accélérer significativement l’électrification de leurs flottes professionnelles dans un avenir proche.
Divertissement
« À la rencontre d’un Hugo : une aventure inattendue »
Le prénom, un véritable reflet de notre identité, peut être à la fois lourd à porter et source de fierté. Dans cette chronique fascinante, le réalisateur Hugo David nous plonge dans son expérience avec un prénom très répandu. Né en 2000, il se retrouve entouré d’autres Hugo, ce qui l’amène à adopter un alias : Hugo D.. Comment ce choix a-t-il influencé son parcours ? Explorez les nuances et les histoires derrière nos prénoms et découvrez comment ils façonnent nos vies dès l’enfance jusqu’à l’âge adulte !

Les Prénoms : Un Voyage au Cœur de l’Identité
Le Rôle Crucial des Prénoms dans nos Existences
Chaque personne possède un prénom, qu’il soit courant ou singulier, et ce dernier peut engendrer à la fois fierté et embarras. Cet article explore la signification profonde et l’influence des prénoms sur notre vie quotidienne. Le réalisateur Hugo David partage son vécu avec un prénom qui a connu une forte popularité durant sa jeunesse.
une Naissance Sous le Signe de la Célébrité
Hugo David est né en 2000 à Tours, une époque où le prénom Hugo était en plein essor. Ses parents, Caroline et Rodolphe, avaient envisagé d’autres choix comme Enzo, également très en vogue à cette période. « Je pense que mes parents ont opté pour un prénom parmi les plus répandus en France plutôt qu’en hommage à Victor Hugo », confie-t-il.
Une Enfance Entourée d’Autres « Hugo »
Dès son plus jeune âge, Hugo se retrouve entouré d’autres enfants portant le même nom. Selon les statistiques de l’Insee,7 694 garçons ont été prénommés Hugo en 2000,faisant de ce prénom le quatrième plus populaire cette année-là. À l’école primaire,il côtoie plusieurs camarades appelés Thibault et autres prénoms similaires. Pour éviter toute confusion lors des appels en classe, les enseignants ajoutent souvent la première lettre du nom de famille après le prénom : ainsi devient-il rapidement « Hugo D. », un surnom auquel il s’habitue sans arduousé.
Pensées sur l’Identité Associée au Prénom
Le choix d’un prénom peut avoir un impact significatif sur notre identité personnelle tout au long de notre existence. Que ce soit pour se distinguer ou pour s’intégrer dans un groupe social spécifique, chaque individu développe une relation particulière avec son propre nom.
les prénoms ne sont pas simplement des désignations ; ils portent avec eux des récits et influencent nos interactions sociales depuis notre enfance jusqu’à l’âge adulte.
-
 Business2 ans ago
Business2 ans agoComment lutter efficacement contre le financement du terrorisme au Nigeria : le point de vue du directeur de la NFIU
-
 Général2 ans ago
Général2 ans agoX (anciennement Twitter) permet enfin de trier les réponses sur iPhone !
-

 Technologie1 an ago
Technologie1 an agoTikTok revient en force aux États-Unis, mais pas sur l’App Store !
-

 Général1 an ago
Général1 an agoAnker SOLIX dévoile la Solarbank 2 AC : la nouvelle ère du stockage d’énergie ultra-compatible !
-
 Général1 an ago
Général1 an agoLa Gazelle de Val (405) : La Star Incontournable du Quinté d’Aujourd’hui !
-
 Sport1 an ago
Sport1 an agoSaisissez les opportunités en or ce lundi 20 janvier 2025 !
-

 Business1 an ago
Business1 an agoUne formidable nouvelle pour les conducteurs de voitures électriques !
-

 Science et nature1 an ago
Science et nature1 an agoDes Projets Ambitieux qui Pourraient Redéfinir la Géopolitique