Général
Détachement de la rétine : un défi persistant pour le syndrome de Stickler !
STOCKHOLM — La détection génétique est essentielle pour diagnostiquer le syndrome de Stickler et identifier les proches à risque. Cependant, elle ne détermine pas le moment idéal pour un traitement rétinien préventif, crucial pour réduire le risque de décollement rétinien. Lors de la réunion annuelle de l’ASRS 2024, le Dr Franco Recchia a souligné l’importance de ces traitements préventifs, qui peuvent sauver la vision des patients. Avec 1 naissance sur 8000 touchée, il est vital d’être vigilant et d’interroger les patients sur leurs antécédents familiaux pour une intervention précoce.

STOCKHOLM – Les tests génétiques, bien qu’utiles pour confirmer le diagnostic du syndrome de Stickler et pour identifier les membres de la famille à risque, ne fournissent pas d’indications sur le moment optimal pour administrer un traitement préventif de la rétine, qui pourrait diminuer le risque de décollement rétinien contralatéral.
Ces résultats ont été présentés lors de la réunion annuelle de la Société Américaine des Spécialistes de la Rétine (ASRS) 2024 par des chercheurs qui ont mené une étude visant à explorer les corrélations entre des mutations spécifiques et la gravité de la maladie.
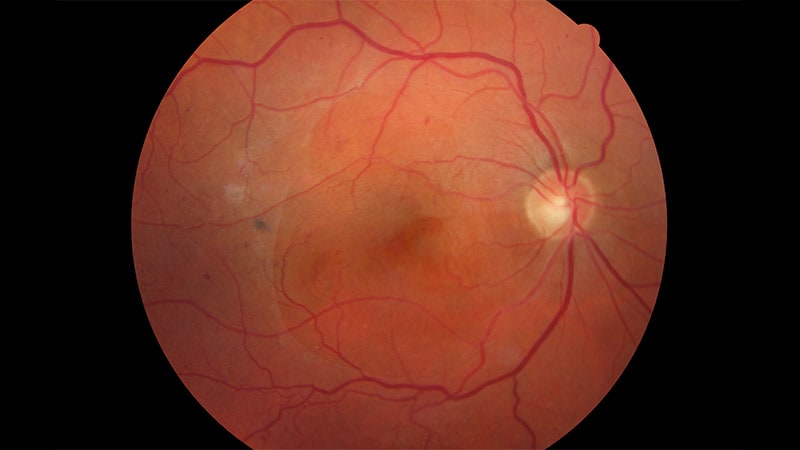
Le syndrome de Stickler touche environ 1 nouveau-né sur 8000. Il est causé par des mutations dans plusieurs gènes, notamment COL2A1 et COL11A1. Ces gènes codent pour des composants du collagène qui confèrent structure et force aux tissus conjonctifs dans les articulations et divers organes. Les mutations dans ces gènes entraînent une variété de signes et symptômes du syndrome, tels que la myopie forte, le glaucome, le décollement de la rétine, la perte auditive et des anomalies squelettiques.

Les décollements rétiniens héréditaires posent des défis particuliers, a déclaré Franco Recchia, MD, spécialiste en vitréo-rétinien chez Tennessee Retina, en rapportant les résultats de l’étude lors de la conférence. Ils touchent généralement des patients plus jeunes et sont souvent bilatéraux.
Recchia a expliqué que les traitements préventifs de la rétine, tels que la rétinopexie prophylactique, pourraient réduire de manière significative le risque de décollement rétinien et de perte de vision chez les patients diagnostiqués. De plus, les membres de la famille identifiés comme à risque pourraient bénéficier d’une intervention précoce pour éviter des complications menaçant la vision.
Analyse de l’Étude
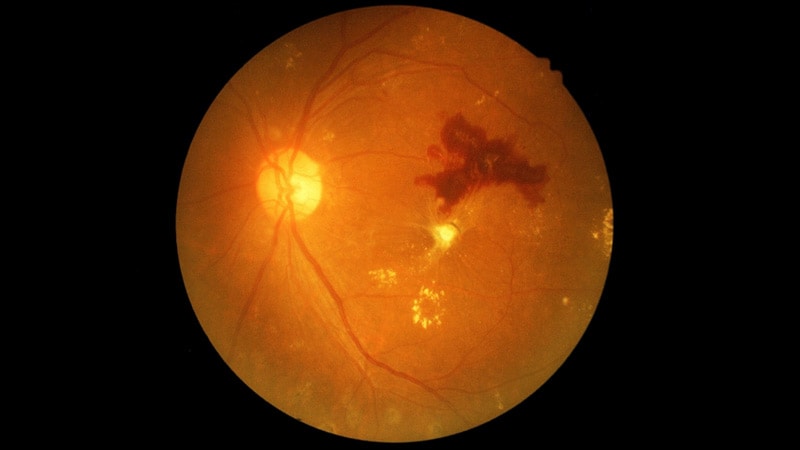
Recchia et son équipe ont effectué des tests génétiques sur 60 individus présentant un décollement rétinien et suspectés d’avoir le syndrome de Stickler, en utilisant une plateforme de test génétique commercialement disponible ciblant les gènes du collagène. L’âge d’apparition du premier décollement rétinien chez ces patients variait de 3 à 46 ans, avec un âge médian de 10 ans.
Les tests ont révélé des variantes pathogènes dans les gènes de collagène COL2A1 ou COL11A1 chez 93 % (56 sur 60) des patients.
En comparant les patients avec les deux variantes, les chercheurs n’ont pas trouvé de différences évidentes dans l’apparition et la latéralité du décollement rétinien ou dans l’incidence de maladies systémiques.
En examinant de plus près la mutation COL2A1, les chercheurs ont constaté que certaines variantes provoquaient une anomalie ou une réduction de la chaîne alpha-1 du collagène de type 2. « Les patients présentant ces variantes particulières avaient tendance à développer la maladie plus tôt et à avoir un décollement plus sévère », a déclaré Recchia.
Les chercheurs ont également pu confirmer que 50 % des membres de la famille de premier degré à risque (13 sur 24) qui ont été testés avaient la maladie, avec des signes cliniques significatifs de glaucome, de perte auditive ou de dysplasie squelettique, mais aucun n’avait de décollement rétinien.
Complexité des Gènes
Plusieurs questions cruciales demeurent sans réponse. « Nous ne savons toujours pas exactement qui traiter, quand traiter, ou même comment », a déclaré Recchia. Il reste difficile de déterminer le moment optimal pour l’intervention, d’établir s’il existe un seuil d’âge au-delà duquel les complications deviennent moins probables, ou de décider si certains enfants nécessitent un traitement précoce plus agressif pour prévenir la perte de vision. « À l’ère de la médecine de précision, nous devrions être en mesure de répondre à ces questions. »
Il a ajouté qu' »il n’y a pas un gène, un effet » chez les patients atteints du syndrome de Stickler. Certaines familles ont des enfants touchés à 5 ans, tandis que d’autres membres de la famille ne présentent rien à 40 ans, ce qui signifie que d’autres gènes pourraient être impliqués.
Michael Gorin, MD, des Instituts Stein Eye & Doheny de l’UCLA, a déclaré que les tests génétiques présentent des limites. Il a expliqué que les tests génétiques pour les gènes associés au syndrome de Stickler chez les patients présentant un décollement rétinien précoce peuvent révéler des variantes d’importance incertaine dans un ou plusieurs gènes. Ces résultats peuvent être difficiles à interpréter, car certaines variantes peuvent être pathogènes, tandis que d’autres sont bénignes. Les variantes de type nonsense ou de codon de terminaison, y compris les décalages de cadre, sont plus susceptibles d’être délétères. Les variantes de type missense sont plus délicates à interpréter. « Dans ces cas, il est nécessaire de mener des investigations supplémentaires avec d’autres membres de la famille informatifs pour évaluer la signification clinique de ces variantes », a-t-il déclaré.

Il a ajouté que les méthodes de test actuelles peuvent ne pas détecter toutes les variantes pathogènes, telles que les variantes introniques ou structurelles. « Ainsi, vous ne pouvez pas vous fier uniquement à la génétique moléculaire, mais vous devez également avoir un bon historique familial et vous appuyer sur des évaluations cliniques plus détaillées du patient et même d’autres membres de la famille. »
Pour surmonter ces limitations, Recchia a déclaré que son équipe de Tennessee Retina élargit l’analyse aux arbres généalogiques familiaux et aux tests multigénérationnels afin de mieux corréler les phénotypes avec les variantes génétiques. Leur objectif est d’élargir leur registre et de créer un consortium multicentrique pour mieux comprendre la maladie.
« Le message principal pour les spécialistes de la rétine est de prendre conscience de la maladie, de demander aux patients leur historique familial et de s’assurer qu’ils ne présentent pas de signes du syndrome de Stickler, car la meilleure chose que vous puissiez faire pour ces patients est de les traiter de manière préventive. »


Général
Anker SOLIX dévoile la Solarbank 2 AC : la nouvelle ère du stockage d’énergie ultra-compatible !
Découvrez le Solarbank 2 AC, une véritable révolution dans le domaine de l’énergie solaire ! Grâce à ses batteries au phosphate de fer lithium, ce système s’adapte parfaitement à vos besoins. Avec une puissance impressionnante de 2400 watts et la possibilité d’ajouter jusqu’à cinq batteries supplémentaires, il assure un stockage optimal. Sa compatibilité avec le compteur Anker SOLIX Smart favorise une gestion intelligente de votre consommation énergétique. Ne ratez pas l’offre spéciale « early bird », disponible dès maintenant pour seulement 999 euros ! Saisissez cette chance unique !

Le Solarbank 2 AC : Une Révolution dans le Stockage Énergétique
Batteries au Lithium Fer Phosphate
Le Solarbank 2 AC se démarque par l’utilisation de batteries au lithium fer phosphate (LFP), reconnues pour leur sécurité et leur efficacité. Ce modèle est particulièrement innovant grâce à son système de couplage alternatif, qui lui permet de s’adapter facilement à divers systèmes solaires déjà en place.Que ce soit pour des installations sur toiture, des systèmes solaires compacts pour balcons ou d’autres configurations réduites, il peut fonctionner avec un micro-onduleur de 800 Watts.
Capacité et flexibilité Énergétique
Avec une capacité maximale d’injection dans le réseau domestique atteignant 1200 watts,le Solarbank 2 AC peut être associé à deux régulateurs solaires MPPT. Cela ouvre la possibilité d’ajouter jusqu’à 1200 watts supplémentaires via des panneaux solaires additionnels, portant ainsi la puissance totale à un impressionnant 2400 watts. Pour les utilisateurs nécessitant davantage de stockage énergétique, il est possible d’intégrer jusqu’à cinq batteries supplémentaires de 1,6 kilowattheure chacune, augmentant la capacité totale à 9,6 kilowattheures.
Intégration dans un Écosystème Intelligent
Le Solarbank 2 AC s’intègre parfaitement dans un écosystème énergétique intelligent grâce à sa compatibilité avec le compteur Anker SOLIX Smart et les prises intelligentes proposées par Anker. cette fonctionnalité permet une gestion optimisée de la consommation électrique tout en réduisant les pertes énergétiques inutiles. De plus, Anker SOLIX prévoit d’étendre cette compatibilité aux dispositifs Shelly.
Durabilité et Résistance aux Intempéries
Anker SOLIX met également l’accent sur la longévité du Solarbank 2 AC. Conçu pour supporter au moins 6000 cycles de charge, cet appareil a une durée de vie estimée dépassant quinze ans. Il est accompagné d’une garantie fabricant décennale et possède une certification IP65 qui assure sa résistance face aux intempéries tout en étant capable de fonctionner dans des températures variant entre -20 °C et +55 °C.
Disponibilité et Offres Promotionnelles
Le solarbank 2 AC est disponible sur le site officiel d’Anker SOLIX ainsi que sur Amazon au prix standard de 1299 euros. Cependant, une offre promotionnelle « early bird » sera active du 20 janvier au 23 février 2025, permettant aux acheteurs intéressés d’acquérir cet appareil dès 999 euros ! Cette promotion inclut également un compteur Anker SOLIX Smart offert pour chaque commande passée durant cette période spéciale.
le Solarbank 2 AC représente une avancée significative dans le domaine du stockage énergétique domestique grâce à ses caractéristiques techniques avancées et son engagement envers la durabilité environnementale.
Business
Une formidable nouvelle pour les conducteurs de voitures électriques !
Excellente nouvelle pour les conducteurs de véhicules électriques ! La recharge gratuite sur le lieu de travail sera exonérée d’impôts jusqu’en 2025. Annoncée par le ministère de l’Économie, cette mesure incitative, en place depuis 2020, s’inscrit dans une dynamique de croissance impressionnante avec une progression annuelle moyenne de 35%. Les entreprises peuvent ainsi offrir des bornes de recharge sans impact fiscal, stimulant la transition écologique. Reste à savoir si cela suffira à convaincre les entreprises hésitantes et à propulser l’électrification des flottes professionnelles vers un avenir durable.

Technologie
Recharge Électrique au Bureau : Une Exonération Fiscale Renouvelée
Les détenteurs de véhicules électriques et leurs employeurs peuvent se réjouir : la possibilité d’effectuer des recharges gratuites sur le lieu de travail sera exonérée d’impôts jusqu’en 2025. Cette décision, annoncée par le ministère des Finances, prolonge une initiative lancée en 2020 pour encourager l’adoption des véhicules électriques dans les entreprises.
Un Secteur en Croissance Dynamique
Cette prolongation intervient à un moment clé, alors que le marché des voitures électriques continue d’afficher une croissance remarquable. Entre 2020 et 2022, la progression annuelle moyenne a atteint 35%. En 2023, les particuliers représentent désormais 84% des acquisitions de véhicules électriques, contre seulement 68% en 2018.
Concrètement,cette mesure permet aux sociétés d’installer gratuitement des bornes de recharge pour leurs employés sans impact fiscal. Les frais liés à l’électricité pour ces recharges ne seront pas pris en compte dans le calcul des avantages en nature. De plus, un abattement de 50% sur ces avantages est maintenu avec un plafond révisé à environ 2000 euros pour l’année prochaine.
Accélération Vers une Mobilité Électrique
Cette initiative fait partie d’une stratégie globale visant à promouvoir l’électrification du parc automobile français. Cependant, les grandes entreprises rencontrent encore des difficultés pour atteindre leurs objectifs ; seulement 8% des nouveaux véhicules immatriculés par ces entités étaient électriques en 2023. Ces incitations fiscales pourraient néanmoins inciter davantage d’employeurs à franchir le pas.Cependant, plusieurs défis demeurent concernant les infrastructures nécessaires au chargement ainsi que sur l’autonomie des véhicules et les perceptions parmi les employés. Par ailleurs, la réduction progressive du bonus écologique pour les utilitaires et sa diminution pour les particuliers pourraient freiner cet élan vers une adoption plus large.
Avenir Prometteur Pour La Mobilité Électrique
Malgré ces obstacles potentiels, il existe un optimisme quant au futur de la mobilité électrique dans le milieu professionnel. Les avancées technologiques continues ainsi qu’un engagement croissant envers la durabilité devraient continuer à favoriser cette tendance vers une adoption accrue des véhicules écologiques.
En maintenant ces mesures fiscales avantageuses jusqu’en 2025 et au-delà, le gouvernement délivre un message fort soutenant la transition écologique dans le secteur du transport. Reste maintenant à voir si cela suffira réellement à convaincre certaines entreprises hésitantes et si cela permettra d’accélérer significativement l’électrification de leurs flottes professionnelles dans un avenir proche.
Divertissement
« À la rencontre d’un Hugo : une aventure inattendue »
Le prénom, un véritable reflet de notre identité, peut être à la fois lourd à porter et source de fierté. Dans cette chronique fascinante, le réalisateur Hugo David nous plonge dans son expérience avec un prénom très répandu. Né en 2000, il se retrouve entouré d’autres Hugo, ce qui l’amène à adopter un alias : Hugo D.. Comment ce choix a-t-il influencé son parcours ? Explorez les nuances et les histoires derrière nos prénoms et découvrez comment ils façonnent nos vies dès l’enfance jusqu’à l’âge adulte !

Les Prénoms : Un Voyage au Cœur de l’Identité
Le Rôle Crucial des Prénoms dans nos Existences
Chaque personne possède un prénom, qu’il soit courant ou singulier, et ce dernier peut engendrer à la fois fierté et embarras. Cet article explore la signification profonde et l’influence des prénoms sur notre vie quotidienne. Le réalisateur Hugo David partage son vécu avec un prénom qui a connu une forte popularité durant sa jeunesse.
une Naissance Sous le Signe de la Célébrité
Hugo David est né en 2000 à Tours, une époque où le prénom Hugo était en plein essor. Ses parents, Caroline et Rodolphe, avaient envisagé d’autres choix comme Enzo, également très en vogue à cette période. « Je pense que mes parents ont opté pour un prénom parmi les plus répandus en France plutôt qu’en hommage à Victor Hugo », confie-t-il.
Une Enfance Entourée d’Autres « Hugo »
Dès son plus jeune âge, Hugo se retrouve entouré d’autres enfants portant le même nom. Selon les statistiques de l’Insee,7 694 garçons ont été prénommés Hugo en 2000,faisant de ce prénom le quatrième plus populaire cette année-là. À l’école primaire,il côtoie plusieurs camarades appelés Thibault et autres prénoms similaires. Pour éviter toute confusion lors des appels en classe, les enseignants ajoutent souvent la première lettre du nom de famille après le prénom : ainsi devient-il rapidement « Hugo D. », un surnom auquel il s’habitue sans arduousé.
Pensées sur l’Identité Associée au Prénom
Le choix d’un prénom peut avoir un impact significatif sur notre identité personnelle tout au long de notre existence. Que ce soit pour se distinguer ou pour s’intégrer dans un groupe social spécifique, chaque individu développe une relation particulière avec son propre nom.
les prénoms ne sont pas simplement des désignations ; ils portent avec eux des récits et influencent nos interactions sociales depuis notre enfance jusqu’à l’âge adulte.
-
 Business2 ans ago
Business2 ans agoComment lutter efficacement contre le financement du terrorisme au Nigeria : le point de vue du directeur de la NFIU
-
 Général2 ans ago
Général2 ans agoX (anciennement Twitter) permet enfin de trier les réponses sur iPhone !
-

 Technologie1 an ago
Technologie1 an agoTikTok revient en force aux États-Unis, mais pas sur l’App Store !
-

 Général1 an ago
Général1 an agoAnker SOLIX dévoile la Solarbank 2 AC : la nouvelle ère du stockage d’énergie ultra-compatible !
-
 Général1 an ago
Général1 an agoLa Gazelle de Val (405) : La Star Incontournable du Quinté d’Aujourd’hui !
-
 Sport1 an ago
Sport1 an agoSaisissez les opportunités en or ce lundi 20 janvier 2025 !
-

 Business1 an ago
Business1 an agoUne formidable nouvelle pour les conducteurs de voitures électriques !
-

 Science et nature1 an ago
Science et nature1 an agoLes meilleures offres du MacBook Pro ce mois-ci !